博文
北京大学夏定国教授最新EER综述|构建下一代锂离子电池全锰基正极材料
|

01 关于这篇文章

文章题目:
Building Better Full Manganese-Based Cathode Materials for Next-Generation Lithium-Ion Batteries
作者:
Jin Song1(宋进), Hangchao Wang1, Yuxuan Zuo1, Kun Zhang1, Tonghuan Yang1, Yali Yang1, Chuan Gao1, Tao Chen1, Guang Feng1, Zewen Jiang2, Wukun Xiao1, Tie Luo3, Dingguo Xia1*(夏定国)
作者单位:
1School of Materials Science and Engineering, Peking University; 2Wuhan University; 3College of Engineering, Peking University
关键词:
Energy storage; Lithium-ion batteries; Cathode materials; Manganese oxides
引用信息:
Jin Song, Hangchao Wang, Yuxuan Zuo, Kun Zhang, Tonghuan Yang, Yali Yang, Chuan Gao, Tao Chen, Guang Feng, Zewen Jiang, Wukun Xiao, Tie Luo, Dingguo Xia. Building Better Full Manganese-Based Cathode Materials for Next-Generation Lithium-Ion Batteries. Electrochem. Energy Rev. 2023, 6(3), 20. https://doi.org/10.1007/s41918-023-00184-8
全文链接:
https://link.springer.com/content/pdf/10.1007/s41918-023-00184-8.pdf

长按/扫描阅读全文
02 图文摘要

03 目录简介

04 综述亮点
1. 回顾了全锰基正极材料的发展历史。
2. 准确描述用于锂离子电池全锰基正极材料的结构、评估其优势及挑战,并讨论解决策略和最新进展。
3. 深入讨论存在争议的热点话题,如在Li2MnO3基正极材料中阴离子氧化还原反应、电压衰减和电压滞后等。
4. 综述关于全锰基正极的新材料及新方法。
5. 从电子-配位-晶体结构视角总结并提出可能增强全锰基材料性能的方案。
05 图文导读
目录简介:

1. 前言
能源使用可大致分为以下三个方面:转换、储存和应用。能源储存是其他二者的桥梁,通过能源储存设备,可实现可再生清洁能源在时间和空间上的便捷使用。在诸多的能源储存设备中,锂离子电池(LIBs)因其高的能量密度、长的循环寿命及其他出色性能,是目前最受欢迎并被广泛应用于日常生活的储能设备。随着便携式电子产品、电动汽车和智能电网的快速发展,需要更为先进的LIBs材料,这些材料不仅应具有优异的性能,还应成本更低。在电池单体级别(Cell-level),LIBs主要由正极材料、负极材料、隔膜和电解液组成,其中正极材料占据所有材料成本的50%左右。这是因为在正极材料中,锂和过渡金属元素更为昂贵,并且正极材料所能提供的实际比容量较低。因此,正极材料在提高实际电池的综合性能方面发挥着重要作用,一致备受关注。
传统的正极材料,如LiFePO4、LiMn2O4、LiCoO2和LiMO2(M = Ni、Co等),在当前社会早已广泛应用。然而,因其较低的能量密度和/或依赖有限的自然资源,难以同时满足对卓越性能和低成本锂离子电池的需求。铁基橄榄石型LiFePO4正极和Mn基尖晶石型LiMn2O4正极,尽管已有商用,但这些化合物理论/实际比容量较低,在追求更高能量密度的文献中早已被替代。钴基或镍基层状正极材料具有超过270 mAh g−1的理论容量。然而,即使在实验室条件下,对于LiCoO2正极材料而言,结构中仅有不到70%的锂离子可以可逆地嵌入/脱出;对于高镍正极,也仅有少于80%的锂离子可以可逆地循环。此外,一旦深度脱锂,正极材料将会发生氧气析出和结构崩塌,引发严重的安全问题,并阻碍其广泛的商业应用。再者,与锰相比,钴和镍经济性不够且生物毒性较高。根据美国地质调查局(USGS)最新数据,全球陆地探明锰储量已经达到15亿吨,远远超过了钴和镍的陆地储量,二者分别约为760万吨和9 500万吨。此外,钴、镍资源分布不均匀。钴储量高度富集在刚果(金,46.05%)、澳大利亚(18.42%)和印度尼西亚(7.89%),这三个地区的钴储量约占全球储量的72%左右。镍储量主要分布在澳大利亚(22.10%)、印度尼西亚(22.10%)、巴西(16.84%)和俄罗斯(7.89%),这四个地区的镍储量约占全球储量的68%左右。然而,全球主要的动力电池制造商主要位于东亚(中国、韩国和日本),他们对上游原材料需求巨大。因此,必须在运输方面投入大量资金,这会增加终端客户的成本。中国现已成为全球最大的锂离子电池生产和消费国。中国动力电池的发展将对全球的“碳中和”进程产生深远影响。然而,中国钴储量(8万吨)和镍储量(280万吨)有限,且品质不高,因此加工难度大。所幸的是中国拥有约5 400万吨的锰储量,一旦全锰基正极材料(FMCMs)在市场上得到广泛开发利用,将会迅速推动新能源产业的进步。因此,开发更好的FMCMs用于下一代锂离子电池似乎是一个令人鼓舞的方向。
与镍、钴相比,锰资源成本和生物毒性较低,近几十年来一直作为潜在正极材料的原材料。近年来,FMCMs还涌现出各种突破,包括一些新结构正极材料的出现和一些新电荷补偿机制的提出。在这,我们系统地梳理了FMCMs的历史,正确描述其结构,评估其优势和挑战,并重点关注相关解决策略和最新进展。特地,本综述还总结了在不同研究尺度上的有效优化方法,并提供一些新的、可能的增强方法,以加快FMCMs的研究进程和实际应用。
2. 用于LIBs的FMCMs的发展历史
图1展示的是FMCMs的发展历史。早在1975年,Ikeda等人就首次提出将二氧化锰(MnO2)用作锂电池正极材料,并使用锂金属作为负极,此刻MnO2/Li电池的放电机制如下:
Li+ + MnO2 → Mn3+O2(Li+)
其中Mn3+O2(Li+)表示Li离子插入MnO2晶格,并且通过将Mn4+还原为Mn3+实现电荷平衡。然而此刻的锂电池被称为锂一次电池,又称锂原电池,是不可再次充电的。

图1 全锰基正极材料的发展历史
可充电锂电池的概念由Whittingham等人在Li-TiS2电池系统中首次提出:以层状TiS2作为正极,锂金属作为负极,电解质为非水电解质。在放电过程中,锂离子首先被插入TiS2层之间的范德华间隙,同时伴随着Ti4+离子还原为Ti3+离子。在充电时,则发生相反的过程:锂离子脱出,同时Ti3+离子氧化为Ti4+离子以维持电荷平衡。在整个充放电(Li离子的脱出/嵌入)过程中,层状结构得到了很好的保持,因此该材料表现出相当好的可逆性。然而,尽管该正极材料可以良好地运行,这个电池系统仍不够可行。由于锂枝晶的产生,并在随后的充放电循环中逐渐生长,最后穿透隔膜,带来严重的爆炸风险。针对该问题,Armand等人在20世纪70年代提出了使用第二种插入式材料取代金属锂的方法,锂离子在两种插入式电极中来回穿梭,这就是“摇椅式”或“穿梭式”电池的来源。紧接着,Goodenough等人提出了一种层状的LixCoO2正极材料,至今仍广泛应用于电池中。受到Fe3O4锂化成LiFe3O4的启发,他们在1983年进一步合成了一种尖晶石锂锰氧化物(LixMn2O4),并将其作为正极材料,在当时就表现出一定的电化学性能,现在仍应用于电动汽车上。
在FMCMs的早期研究阶段,人们对开发新型锂锰氧化物表现出极大的兴趣,因此许多研究团队合成了各种不同相结构的锂锰氧化物。Thackeray等人在1991年通过酸刻蚀从岩盐相Li2MnO3(Li2O·MnO2)中脱出Li2O,开发出一种Li2−xMnO3−x正极材料,其可逆容量约为200 mAh g−1。与此同时,合成具有类似于LiCoO2的二维锂离子通道结构的层状LiMnO2,是一个长期以来的目标。然而,单斜层状LiMnO2(m-LiMnO2)在热力学上并不稳定,正交结构的o-LiMnO2(锯齿状层状结构)更为稳定,尽管这二者稳定性差异非常小。相较于o-LiMnO2,m-LiMnO2得到更多研究者的关注,如前所述它的结构与LiCoO2非常相似,更有可能成为实际的电极材料。部分研究团队已经合成制备了层状m-LixMnO2材料,然而这些有趣的材料通常是水合的、含有质子或是非化学计量比的。1996年,Bruce等人首次报道了无水和满足化学计量比m-LiMnO2的合成方法,该方法通过从NaMnO2前驱体中进行Li/Na离子交换获得层状m-LiMnO2。该材料显示出相当高的充电比容量(270 mAh g−1)以及较为良好的循环稳定性。随后,Tabuchi等人也独立报道了层状m-LiMnO2的水热合成。在本世纪初期,模板法也是一种极具吸引力的层状LiMnO2的合成方法,其可以有效控制制备得到的纳米晶体的尺寸。另一种在热力学上稳定的FMCMs是层状单斜的Li2MnO3(或Li[Li1/3Mn2/3]O2),Mn层中1/3的Mn离子被Li离子取代,形成Li-Mn-Mn超晶格,并带来了C2/m对称性。由于Mn层中存在过量Li离子,以氧为配位中心,将出现“Li–O–Li”构型,形成了孤立的O态(未杂化的O 2p态或非成键的O 2p轨道),其能量高于成键O态,使得Li2MnO3体系中阴离子O参与电荷补偿成为可能,因而在理论上可能实现更高的能量密度。然而,Li2MnO3通常只有通过纳米化或是在高温条件下进行测试,才表现出相对可观的电化学性能。此外,由于Mn4+难以被氧化到更高的价态(这一点存在争议,后面将进一步讨论),在活化过程中,纯阴离子O参与氧化反应,将释放出大量氧气,导致不可逆的结构转变和糟糕的循环性能。
在接下来的数十年中,FMCMs的发展并没有取得显著进展,大多数FMCMs可以看作是上述结构的组合,如Li2MnO3·LiMnO2、Li2MnO3·LiMn2O4等。近年来,考虑到环境问题和电动汽车的快速发展,越来越多的研究人员再次研究FMCMs,并成功合成了一些引人注目的FMCMs,这些材料展示出非凡的电化学性能。在2014年,Ceder等人将渗流理论引入到LIBs中,扩展了正极材料的设计空间。随后,诸多阳离子无序岩盐结构FMCMs和类尖晶石结构部分离子有序的FMCMs被创造性地开发出来,并通过多电子氧化还原实现超高的能量密度,呈现出作为正极材料的巨大潜力。此外,通过更加关注过渡金属层内阳离子的有序性,一系列具有不同层内金属/空位/过渡金属(M/V/TM)排列的FMCMs被设计合成,从而建立起层内离子排列、结构响应和阴离子可逆性/稳定性之间的关联。更为有趣的是,通过晶体结构对称性和/或界面轨道有序性,可以很好地控制Mn3+的Jahn-Teller畸变。一些研究团队还提出并合成具有不同氧亚晶格的FMCMs。O2构型的FMCMs因为过渡金属离子难以迁移到Li层的八面体位,不会发生层状-尖晶石相变,因而没有明显的电压衰退现象。此外,大多数FMCMs主要基于体相中的氧化还原反应,受限于电极中较为缓慢的锂离子扩散过程,倍率性能存在不足。因此,有研究团队专注于表面氧化还原反应,合成了一些有趣的FMCMs,并提出一些非常规的电荷补偿机制。这些新材料和新概念给予研究人员极大的信心和灵感,对于FMCMs的研究具有重要意义,极大地拓展了正极材料的设计和开发空间。
3. FMCMs的优势
FMCMs因其诸多显著优势而备受关注。考虑到成本和电化学性能,正极材料早已被证明是构建更好电池的瓶颈所在。图2a显示的是在电池单体级别(Cell-level),电池制造成本和材料成本的分布情况。材料成本占总制造成本的主要部分,约占总制造成本的75%,其中正极材料约占所有材料成本的50%左右(图2a,左上角)。为了实现更具吸引力的成本结构和利润率,低成本和高能量密度正极材料的获取至关重要。图2b总结了过去两年中三种常见过渡金属的价格变化趋势。钴的价格波动较大,镍的价格逐渐上涨,锰的价格则相对稳定。钴化合物的高成本和毒性促使开发钴基正极材料的替代材料。由于具有相对较高的比容量和良好的稳定性,高镍正极材料早已得到广泛研究并逐渐应用于市场,但单体电池价格未达预期,且还面临氧气析出和合成难度等问题。此外,随着电动汽车和储能电网的大规模普及,预计镍和钴资源的价格和消耗率将进一步增加。有研究表明,一旦锂离子电池产业的年产量达到1 TWh,将需要约100万吨钴或镍的化合物,从而对金属资源造成巨大压力。与钴、镍价格相比,锰的价格远低于钴的价格,不到镍价格的四分之一,如图2b所示。
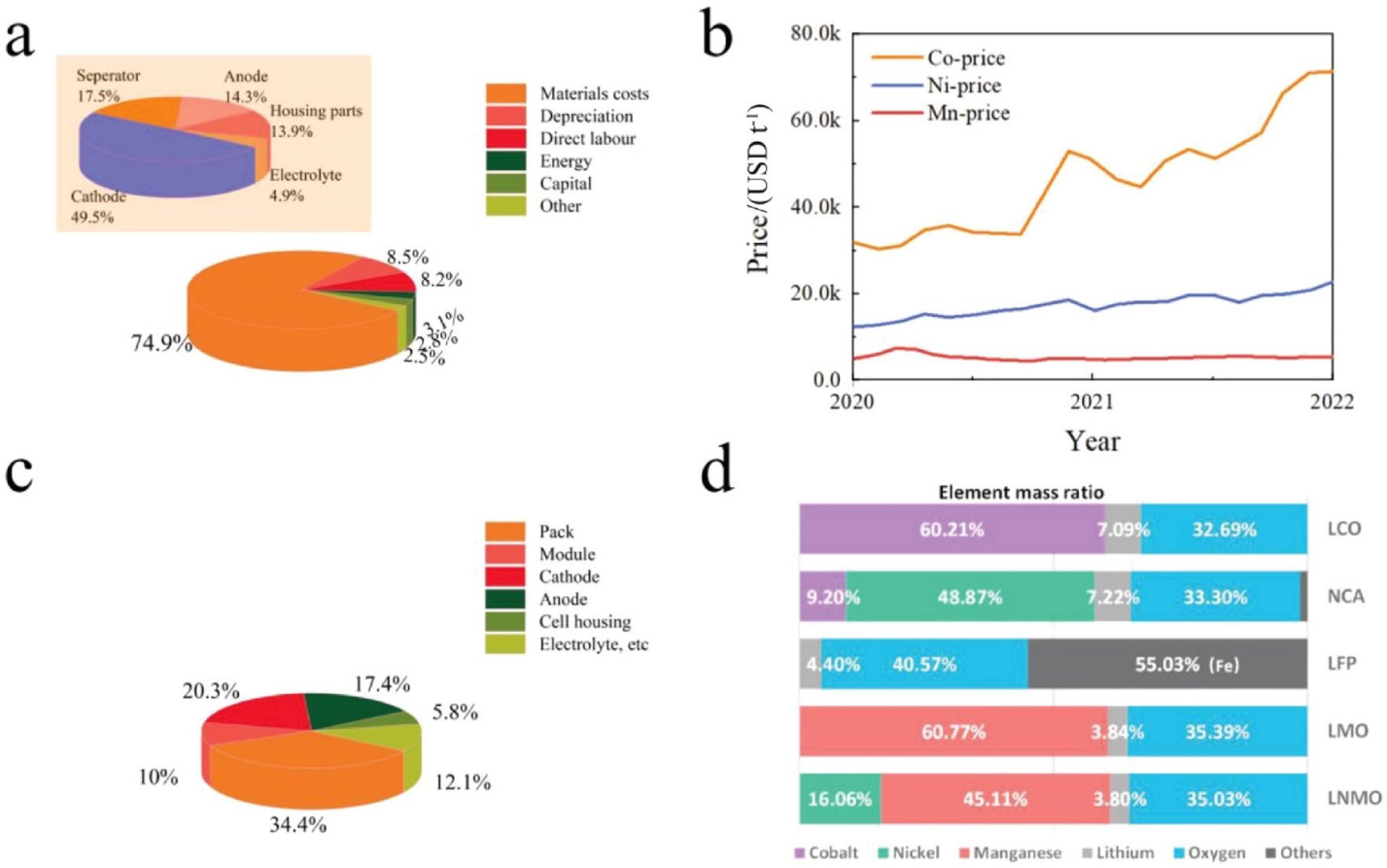
图2 a 电池单体级别(Cell-level)的制造成本分布和材料成本分布。b 近两年锰、钴、镍的价格变化(来源:https://tradingeconomics.com/)。c 电池系统质量分布。d 常见正极中元素质量占比。Copyright 2020, Elsevier
另一方面,电池模组中电极材料的质量比接近40%,正极材料占总质量的20%以上,如图2c所示。随着对电池系统的进一步优化,比如比亚迪的刀片电池、宁德时代的CTP(Cell to Pack)技术等,活性物质的质量占比将进一步提高。图2d展示的是几种常见正极材料的元素质量占比,对于层状氧化物材料,钴和镍占据了很大比例。因此,通过锰取代钴、镍,构建高能量密度、低成本的全锰基正极材料,将是一个很好的替代方案,这将显著降低电池成本,有力促进电动汽车轻量化或长续航的快速发展。
此外,锂锰氧化物及其衍生物具有多种成分和结构,能够被用于,事实上已有部分被用于锂离子电池正极材料。这些FMCMs基于锰离子的氧化还原反应,因而具有相对较高的氧化还原电位,与当前的电解液系统兼容。在一些富锂或锂过量的FMCMs中,阴离子氧的氧化还原反应也可以发生,进一步提高材料的实际放电容量。令人惊讶的是,一些FMCMs甚至可以提供超过350 mAh g−1的超高可逆容量,远高于镍或钴基正极。锰离子属于多变价离子,还可以将锰离子的氧化还原电对控制在Mn2+到Mn5+甚至到Mn7+之间。有研究表明,在过充时,FMCMs也更加安全。毫无疑问,FMCMs是下一代锂离子电池正极材料的潜在候选者。
4. 常见FMCMs的结构
如上所述,锂锰氧化物的结构化学非常复杂。所幸的是,现如今已成功合成了多种晶体结构的锂锰氧化物,并进行了详细探索,这为正极材料的研究提供了广泛的选择。在这里,我们更加关注应用于锂离子电池领域的FMCMs的结构,并对它们进行准确描述,如图3。
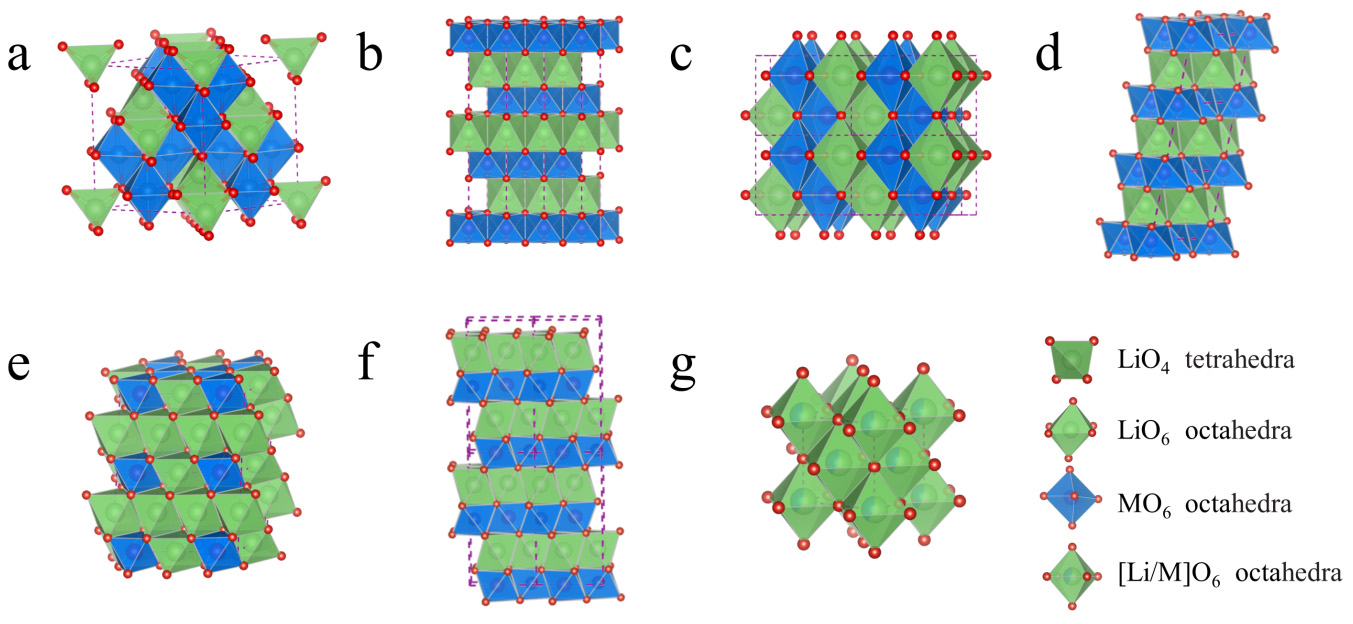
图3 部分重要FMCMs的结构:a 尖晶石LiMn2O4(Fd(3–)m);b 层状LiMO2(R(3–)m);c 锯齿状LiMnO2(Pmnm);d 层状LiMnO2(C2/m);e 层状Li2MnO3(C2/m);f O2构型LiMnO2(P63/mc);g 阳离子无序锂锰氧化物(Fm(3–)m)
4.1 尖晶石LiMn2O4
LiMn2O4是典型的尖晶石结构正极材料,其理论容量为148 mAh g−1,是最容易制备得到的锂锰氧正极材料之一,目前已在商业锂离子电池中得到应用。在尖晶石结构中,氧离子以面心立方密堆积的方式排列,而阳离子则排列在O密堆积的间隙位,如图 3a。可用Fd(3–)m空间群对尖晶石结构进行标定,面心立方O的Wyckoff位点为32e,Li和Mn离子分别占据了四面体8a和八面体16d位,八面体16c位则处于未占据态,因此LiMn2O4的原子分布可以表示为[Li1](8a)[Mn2](16d)[O4](32e)。此外,四面体8a位和未占据的八面体16c位相互共面,呈现共边八面体阵列,从而形成稳定的锂离子三维通道,允许锂离子快速扩散。因此,立方尖晶石结构正极材料表现出优异的倍率性能。
4.2 层状LiMnO2
对于理想化学计量比的LiMnO2,有两种常见的结构被用作正极材料,层状结构(m-LiMnO2)和正交结构(o-LiMnO2)。m-LiMnO2的晶体结构与LiMO2(M = Ni、Co等)类似。LiMO2结晶成完全有序的α-NaFeO2菱方层状结构,属于R(3–)m空间群,其中O离子堆积方式和尖晶石结构中的一样,均为面心立方密堆积。Li和M离子则位于面心立方密堆积的八面体间隙位,交替占据(111)面,沿c轴形成O-Li-O-M-O层序,从而产生ALiBMCLiAMBLiC阵列(大写字母:A、B、C,代表结构中的氧亚晶格;下标Li和M分别表示Li层和M层),如图3b所示。每个单胞由M离子和O离子形成3个刚性的MO2(−)层,因此该结构被标定为O3构型(这里的O指的是八面体)。在该结构中,锂离子在锂层是可移动的,因此,锂离子可以可逆地从锂平面中脱出/重新插入,形成快速的二维锂离子扩散通道;此外,共边的MO6八面体排列能提供直接的M–M相互作用,此类结构材料通常表现出良好的电子导电性。因此,O3构型的层状LiMO2氧化物一直是富有吸引力的正极材料,如LiCoO2、三元正极、高镍正极等。层状的m-LiMnO2也是一种O3构型的氧化物,但由于高自旋Mn3+离子Jahn-Teller变形的Mn3+O6八面体的协同扭曲而表现出晶格的单斜畸变,因此其属于C2/m空间群而不再是R(3–)m空间群,如图3d所示。此外,m-LiMnO2的热力学稳定性不如o-LiMnO2,通常只能通过软化学方法合成。o-LiMnO2也具有有序的岩盐结构,空间群为Pmnm,如图3c所示。有趣的是,LiO6和MnO6八面体排列在波纹层中,因此也可称之为锯齿状层状LiMnO2。
4.3 层状Li2MnO3(或Li[Li1/3Mn2/3]O2)
层状Li2MnO3(或Li[Li1/3Mn2/3]O2)的氧堆积方式与LiMO2类似,如图3e所示。但过渡金属层不再是仅含过渡金属离子的层。在模拟的单胞中,可看成是晶胞中25%的Li取代了33%的Mn总位点(Wyckoff位置:2b),形成阳离子有序的Li/Mn混合层(Li1/3Mn2/3或LiMn2)。这种有序排列和Li、Mn离子半径的差异使其对称性从R(3–)m对称降低为C2/m对称。其化学式可写成Li1.0(4h)Li0.5(2c)[Li0.5(2b)Mn(4g)](O1)4i(O2)8j。
4.4 O2构型FMCMs
另一种层状FMCMs是O2构型的层状锂锰氧化物。1999年Dahn以P2构型的Na2/3MnO2为前驱体合成了O2构型的Li2/3MnO2。Delmas等人也在2001年首次报道了一种具有独特ABCBAB氧堆积的O2构型LiCoO2,与具有ABCABC氧堆积的层状O3构型LiCoO2相比,O2构型LiCoO2是处于热力学亚稳态的。这里O2构型中“O”的含义与前面提到的O3构型层状结构中的“O”相同,指的是八面体,这里的“2”是指描述材料单胞所需的最少MO2(−)层数。O2型结构中LiO6八面体和MO6八面体的连接形式与O3型结构中的连接形式不同。在O2构型中,LiO6八面体与MO6八面体共用边和面,如图3f,而在O3构型中,LiO6八面体与MO6八面体仅共用边。更为重要的是,M离子很难从M层的八面体位置通过四面体位置迁移到Li层的八面体位置,即使M离子可以迁移到四面体位置,迁移后的M离子与共面的M离子之间存在巨大的排斥作用,将会阻碍进一步的迁移,因此O2构型正极材料在常规条件下几乎不可能发生层状到尖晶石的转变。
4.5 O2新型FMCMs
近年来,随着渗流理论的引入,Ceder等人报道了一种新型的正极材料结构—阳离子无序结构。阳离子无序结构仍属于岩盐结构,其空间群为Fm(3–)m,拥有类似于层状结构中氧的面心立方密堆积框架,但Li和过渡金属离子的排列方式存在差别。层状结构中Li和过渡金属离子交替占据(111)平面,而在阳离子无序结构材料中,Li和M离子则随机分布在金属层(锂层和过渡金属层)中,即所谓的阳离子无序,如图3g所示。
5. FMCMs面临的问题和挑战
5.1 共性问题
尽管FMCMs因其显著优点而具有相当的吸引力,但由于Mn离子的电子、配位结构和电极晶体结构,仍然存在各种问题和挑战,大大阻碍了FMCMs在市场中的普及。大多数常见过渡金属氧化物正极材料中六个配体围绕其中心过渡金属离子对称排列,呈现八面体(Oh)配位。根据配体场理论,八面体环境下的五个3d轨道分裂为两个简并轨道,通常为t2g轨道(3dxy、3dyz和3dxz)和eg轨道(3dx2−y2和3dz2)。然而,由于高自旋态的Mn3+离子,其电子分布为t2g3eg1。在含高自旋Mn3+的锂锰氧化物中的配位场会使得六个等效的M–O键发生畸变,形成四个较短的赤道平面键和两个较长的轴向键,这进一步分裂d轨道能级。这种现象被称为Jahn-Teller畸变,涉及在晶体场中部分消除离子的d电子简并性,从而降低晶格对称性。协同的Jahn-Teller畸变进一步影响材料晶体结构的对称性,这可以很好地解释为什么理想的R(3–)m LiMnO2不存在;而对于尖晶石结构的LiMn2O4,电极在持续循环过程中,无法保持结构完整性。这也是尖晶石LiMn2O4材料在通过3.0 V区域时,容量快速衰减的重要原因
另一个共有问题是Mn离子的溶解问题。这是大多数锰基正极材料面临的极具挑战性的问题。它直接损害晶体结构的完整性,极大地降低了电池的容量和循环性能;此外,溶解的Mn离子通过液态电解质发生迁移并沉积在负极材料表面,对固体-电解液界面(SEI)膜造成严重损害,进一步降低电池的电化学性能。尽管在这一领域进行了诸多研究,但关于Mn离子的溶解机制尚无共识。根据之前的研究,目前有两种较为主流的解释:i)Mn3+的歧化(2Mn3+ → Mn4+ + Mn2+),Mn2+可在溶解电解液中;ii)电解液中酸性物质腐蚀正极材料,导致Mn离子溶解。这两种机制都与Jahn-Teller畸变相关,Jahn-Teller畸变可能会促进Mn3+的歧化和/或增加与酸(如LiPF6水解而在电解液中生成的HF)的反应性,从而增加LIBs中Mn离子的溶解。然而,也有研究小组发现,随着电极被过度充电,溶解的Mn2+离子浓度也迅速增加。最大溶解发生在充电结束时,此时Mn离子的氧化态为4+,这意味着存在除Mn离子歧化以外的其他重要机制。脱锂态的本征不稳定性导致MnOx的流失,形成更稳定的单相结构,这也是不能忽视的。此外,高活性脱锂态MnOx与电解液之间的界面反应也值得关注。这些具有挑战性的问题几乎存在于所有FMCMs中,并且对FMCMs的结构演化和电化学性能产生显著影响,需要深入研究和解决。
5.2 尖晶石LiMn2O4的问题
尖晶石LiMn2O4室温下可以在4.0 V范围内进行良好的循环。完全脱锂状态下,Mn2O4仍保持尖晶石结构,且因为强共边八面体Mn2O4阵列,Li离子可以在四面体位点进行可逆的嵌入/脱出,而不破坏尖晶石的三维框架。然而,当在高温下进行循环时,其性能会快速下降,通常归因于Mn离子在电解液中的溶解、高电压下电解液的分解以及在锂化状态下因Jahn-Teller畸变引起的不可逆相变。上述性能下降的原因并不孤立,更可能是所有因素的协同:Jahn-Teller畸变促进了Mn3+的歧化,增加与酸的反应性,而Mn4+则可能攻击电解液并引起电解液的分解。当放电到较低电压(3.0 V)时,额外的Li离子可以嵌入空的16c八面体位,实现双倍容量的释放。然而,位于8a四面体位点的Li离子与位于16c八面体位点的Li离子共面,因此强烈的静电斥力将导致8a四面体位点上的Li离子向相邻的空的16c八面体位点迁移,从而形成具有新离子分布的有序岩盐结构,即[Li2](16c)[Mn2](16d)[O4](32e)。从8a到16c位点Li离子的集体迁移,是嵌锂过程中出现一级相变和低电压平台的原因,增加在实际应用时,电池管理的难度;此外,由于尖晶石框架的破坏,LiMn2O4电极在经过3.0 V时,循环性能差。同时,Li离子的扩散路径受到Jahn-Teller畸变和四方相与立方相晶格不匹配的阻碍,导致循环过程中动力学缓慢和容量快速退化。
5.3 LiMnO2的问题
在LiMnO2中,所有Mn离子价态均为3+价,高自旋的Mn3+离子带来严重的Jahn-Teller畸变。在上述LiMnO2的两种结构(m-和o-LiMnO2),因协同Jahn-Teller效应,室温下阴离子O的亚晶格从理想的立方密堆积发生扭曲。根据合成条件,它们可以进一步分为两类:低温(LT)m/o-LiMnO2材料和高温(HT)m/o-LiMnO2材料。LT o-LiMnO2可以在4.0或2.8 V电压平台上实现相对较好的循环稳定性,然而仅有大约一半的理论容量可以被释放,这有违寻求高能量密度的目的。在两个平台上循环时,LT o-LiMnO2可提供相对较高的比容量(190 mAh g−1),然而,在长时间循环中,容量保持率较差。HT o-LiMnO2通常在较宽的电压范围内显示出比LT相更好的稳定性。然而不幸的是,几乎所有的LiMnO2在循环过程中都会经历严重的层状-尖晶石相变。此外,研究还发现将o-LiMnO2材料充电至更高电压(更深的脱锂状态)时,会加快这种转变的速度,这限制了可利用的实际容量。m-LiMnO2因其结构与成功商业化的LiCoO2正极材料高度相似,且锰的成本远低于钴,其具有很高的商业和科学价值。m-LiMnO2在热力学上不是一个稳定的结构,因此几乎不存在HT m-LiMnO2。理想化学计量比的m-LiMnO2是由“软化学”方法(锂/钠离子交换法)首次制备得到。紧接着,Tabuchi等人也独立报道层状m-LiMnO2的另一种 “软化学”方法合成(水热合成)。通过“软化学”方法制备的m-LiMnO2,在这命名为LT m-LiMnO2,可释放相当客观的比容量(> 200 mAh g−1)。然而,这些材料的循环性能不够好,随后的放电曲线显示出快速的容量下降和持续的层状-尖晶石相变。Ceder等人提出,脱锂态的LixMnO2由层状向尖晶石的转变有两个过程。具体而言,在第一阶段,当材料部分脱锂时,相当一部分Mn离子迅速迁移到被锂空位包围的四面体位置,并形成亚稳态中间体。计算得出的活化势垒较低,部分原因是由于Mn离子迁移到四面体位点得到Mn3+歧化(2Mn3+oct → Mn2+tet + Mn4+oct)的促进。在第二阶段,四面体的Mn离子和剩余的八面体位的锂离子进行协调重排,形成最终的尖晶石相。这个过程速度较慢,主要是因为该过程更加复杂且具有更高的活化势垒。层状-尖晶石相变直接影响电压曲线特征,LiMnO2材料会出现明显不同于其他层状LiMO2(M = Ni、Co等)正极材料的双电压平台,增加电池状态监测的难度。
5.4 Li2MnO3和Li2MnO3基材料的问题
层状Li2MnO3是另一种重要的FMCMs,其理论容量为458 mAh g−1。一般而言,在常规条件下,结晶性良好的Li2MnO3不会显示任何的电化学活性;然而,在较高温度下进行循环时,其将能提供可观的可逆容量。此外,具有氧空位、层错缺陷或纳米结构的Li2MnO3也展示出较为明显的电化学活性。理解Li2MnO3及其他Li2MnO3基材料异常首次充电容量的起源是一个具有挑战性的问题,目前仍存在争议。有研究表明,Li2MnO3中的Mn4+离子难以被氧化为Mn5+;但一些研究团队则认为,在高电位下,伴随着Mn离子迁移到四面体位,可能发生Mn4+/7+的氧化还原反应。当然,这不能被排除,因为Mn7+离子可能处于亚稳态或作为反应中间体参与了O2−离子的氧化反应,进而形成困在颗粒中的分子O2,难以表征。
Ceder等人于2016年指出,富锂正极材料相较于常规的非富锂层状材料,由于在过渡金属层内引入Li,若以O为中心原子,便会出现“Li4M2-O”的氧配位和“Li–O–Li”构型,如图 4a、b所示。再由配位化学可知,沿着“Li–O–Li”构型的O 2p轨道将是非杂化的O 2p态,其在能量上高于键合的O 2p态,因此更容易被氧化。沿着“Li–O–Li”构型上的非杂化O 2p态不稳定电子的电荷补偿,是富锂锰基正极材料表现出超过基于过渡金属氧化还原理论容量以外容量的根源,目前已被广泛接受。但是,关于阴离子O氧化物种的具体形式出现了新的争议。Bruce等人指出,单线态O2可以在高充电电压下产生,并与电解液相互作用生成CO2。一旦阴离子氧发生配位结构缺失,氧损失将主导氧的演变。对于结晶性良好的Li2MnO3,他们利用原位电化学质谱(图4c)和6Li 核磁共振谱图定量地证明,在第一次充电过程中,锂离子的脱出仅通过粒子外壳的氧损失(O-loss)来进行电荷补偿,而不是通过在晶体框架内O2−的氧化还原(O-redox)。Zhou等人也证实了这一结果,他们还强调,近表面区域的表面碳酸盐物种分解和氧气释放的结合,是Li2MnO3首次充电时出现电压平台的原因。然而有趣的是,也有研究团队发现在Li2MnO3中存在部分可逆的阴离子氧化还原反应。富锂锰基正极材料在阴离子O的氧化还原可逆性方面与纯Li2MnO3存在显著差异。对于富锂锰基正极材料,Bruce等人通过结合各种先进的表征技术,如共振非弹性X射线散射光谱(RIXS,图4d)、软X射线吸收光谱(sXAS)、拉曼光谱和X射线吸收近边缘结构光谱等证明,除氧损失外,锂离子的脱出还通过在氧上产生局域电子空穴进行电荷补偿,而不是形成真正的O22−二聚体。通过RIXS和17O魔角自旋核磁共振谱,他们进一步表征到分子O2,如图 4e–g所示,在充电到4.6 V时,分子O2出现在富锂锰基正极颗粒内,并在放电到3.75 V的较低电压下时可还原为O2−。与之不同的是,Zhou等人通过原位技术,如图4h所示的拉曼光谱和XRD技术对一种典型的富锂锰基正极材料Li1.2Ni0.2Mn0.6O2进行表征,他们发现,O−-O−过氧化物二聚体能够在高电位形成并且能可逆地发生O2−/O−的氧化还原过程。有趣的是,课题组等人发现阴离子O氧化还原响应形式,可以通过改变过渡金属层内阳离子排列进行氧离子局部对称性操控进而发生改变。在氧离子局部对称性改变的Li2RuO3富锂模型材料中,氧的电荷补偿是以O-Ru-O伸缩形式进行响应,而不再是O−-O−的二聚体反应形式,并实现稳定的电池循环。总的来说,关于Li2MnO3和Li2MnO3基正极材料的电荷补偿机制仍存在一些谜团和争议,需要进行更多的研究,尤其是关于氧化O物种的具体形式及其在循环过程中的演化。另一方面,无论是纯Li2MnO3还是富锂锰基正极材料,都存在氧损失:大量阴离子O的氧化还原反应导致O2或CO2气体的产生(可通过差分电化学质谱进行表征),从而导致不可逆的结构转变、表面致密相的形成以及电化学活性的降低。
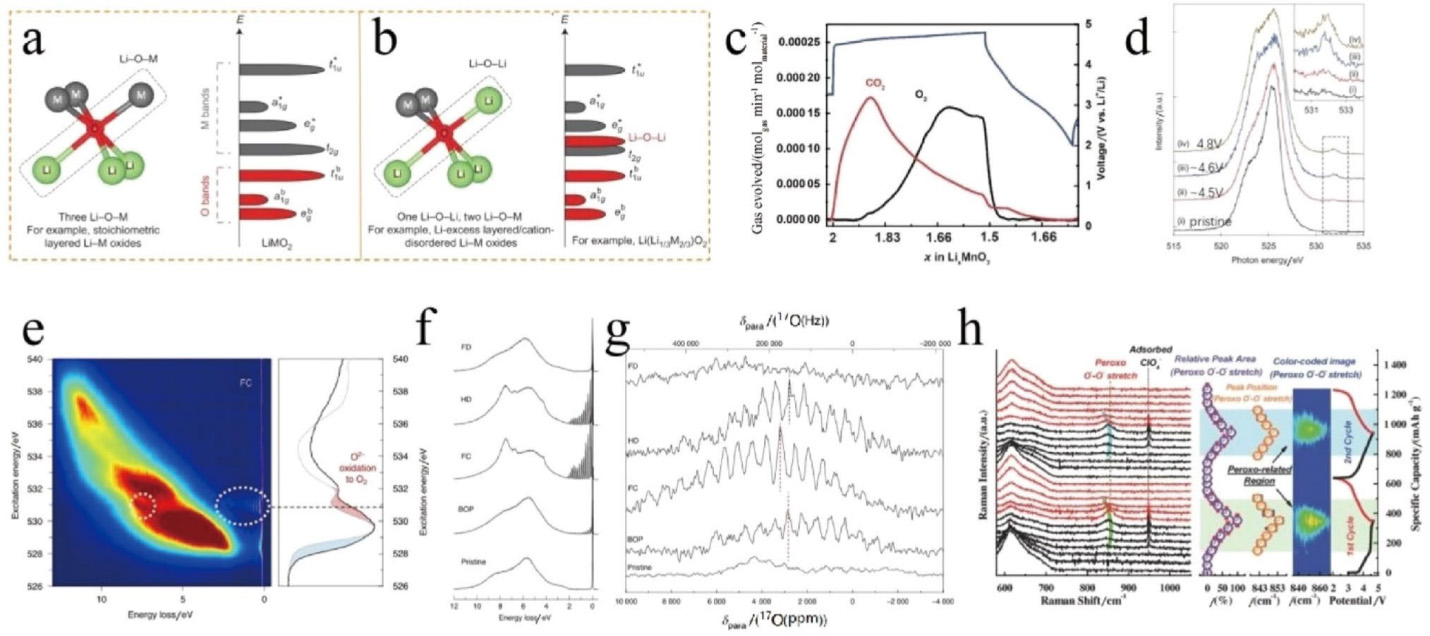
图4 富锂锰基正极材料中阴离子电荷补偿的起源及氧化O的物种形式。a 非富锂环境下,O的配位情况以及对应的能级分裂情况;b 富锂环境下,O的配位情况以及对应的能级分裂情况,出现了新的“Li–O–Li”构型对应的非杂化的O 2p态。Copyright 2016, Nature。c 在Li2MnO3中,原位差分电化学质谱证实氧化O以氧损失形式存在,包含氧气和二氧化碳。Copyright 2020, American Chemical Society。d 通过RIXS证明氧化O以在氧上产生局域电子空穴形式存在。Copyright 2016, Nature。e–g 证实氧化O以分子O2的形式存在。Copyright 2020, Nature。h 通过原位Raman证实氧化O以O−-O−二聚体的形式存在。Copyright 2018, Wiley-VCH
此外,Li2MnO3具有相当大的带隙且阴离子O氧化还原迟缓,导致较差循环性能,使其在工业上不具吸引力。因此,研究人员更加关注Li2MnO3基的材料,主要是富锂锰基正极材料(Li2MnO3*LiMO2,M = N、Co、Mn等)。由于引入含Ni、Co等活性元素的层状LiMO2,因此其表现出更好的电化学性能。富锂锰基正极能够通过将过渡金属阳离子的氧化还原反应与阴离子O的氧化还原反应相结合,释放更高的比容量,并具有良好的循环稳定性,而不是像在Li2MnO3中,进行纯粹的阴离子O的氧化还原反应。然而,在后续循环过程中,富锂正极出现明显的电压衰退现象。与其他正极材料相比,电压衰退是富锂正极面临的独特问题。它带来的主要不利影响是使得构建有效的电池管理系统变得困难;此外,随着电压衰退,实际输出能量密度持续降低,因而极大地阻碍了富锂正极的商业应用。
Li2MnO3基正极材料面临的另一个挑战是研究较少的电压滞后问题。电压滞后会造成巨大的能量损失,可能会以热量的形式耗散,从而给终端用户带来额外的能量成本,这也会使得电池的热管理和状态监测复杂化。一般来说,电压滞后可以大致理解为在相同Li含量下充放电电压偏离平衡电位的现象,其有两个来源:动力学和热力学来源。源自动力学的电压滞后,即所谓的过电位,可以通过降低电荷传输过程中的电阻或者降低电流密度来降低,当电流降低到足够小时,过电位可以消失。热力学起源滞后通常是路径相关的,起源于充放电过程中不对称的反应路径,因此不同吉布斯自由能对应的中间相的充放电电压值不同,即热力学起源导致的滞后特性。早期研究人员发现,Li2MnO3基正极材料首圈电压曲线与后续循环电压曲线明显不同。4.5 V的电压平台和较低的首次库伦效率可通过Li2MnO3组分的激活,伴随者阴离子O的氧化还原、氧损失以及离子重排来解释。然而,关于离子重排的更多细节仍不清楚,并且仍然没有直接的表征用来量化结构响应和性能曲线之间的关系。此外,令人困惑的是,GITT结果显示,在Li2MnO3组分被激活的富锂锰基正极材料中,即使经过充分的驰豫,充放电电压仍旧不能回到相同的平衡电位,电压差约为200 mV。这与常见的层状LiMO2(M = Co、Ni等)正极材料或未激活Li2MnO3组分的富锂锰基正极材料存在显著差异。更为重要的是,研究人员还观察到了一些有趣的现象:一般说来,当加热电极系统时,固体基质中缓慢的Li离子扩散和迁移过程可能被加速。然而,Ohzuku等人发现,在55 ℃下观察到富锂锰基正极材料的电压迟滞曲线比室温条件下更大。这表明Li2MnO3基富锂正极材料中的电压滞后不可忽视,这与其他传统层状正极有着明显差异,且更为复杂。Li2MnO3基富锂正极材料中电压滞后的成因及其解决方法亟待研究。
6. 改进策略与研究进展
6.1 针对LiMn2O4基正极材料
如上所述,尖晶石LiMn2O4经历低电压放电时,将出现两个电压平台、容量快速退化和倍率性能差的问题。因此,当前用于商业正极的尖晶石LiMn2O4主要在4.0 V电压范围内循环使用。然而,在高温测试条件下,仍旧容易发生明显的容量衰减,这主要与Mn离子Jahn-Teller畸变和/或歧化反应引起的Mn离子溶解密切相关。因此,能够抑制Mn离子溶解的方法均被提出用以提高电极性能。根据LiMn2O4的化学式,其中一半的Mn离子是高自旋的,因此,采用单价阳离子(主要是Li+)或二价阳离子掺杂(Mg2+、Zn2+等)等策略来提高Mn离子的平均价态是有效的。当然,三价阳离子掺杂也是常见且有效的方法,可以分为两个部分,即非活性三价阳离子掺杂或活性三价阳离子掺杂。Al3+作为一种非活性掺杂离子,较早被广泛应用。计算得到的Al–O键比Mn–O键强得多,因此Al3+取代可能抑制尖晶石结构在锂离子嵌入/脱出过程中的膨胀或收缩。Ga、La等也被用于改善电极性能,然而,由于这些离子没有电化学活性,通常实际放电容量会降低。Cr、Co、Ni等活性离子也被用作掺杂离子。Cr掺杂材料通过增强Cr–O键,抑制局部畸变(MO6八面体)和减小体积收缩来提高循环稳定性。Co离子和Ni离子部分取代LiMn2O4中的Mn离子,使得Mn4+离子的比例增加,高自旋Mn3+离子引起的Jahn-Teller畸变将被抑制,因而其容量保持率优于未掺杂的尖晶石。而更多Co或Ni离子的掺杂将构建高电压尖晶石正极材料,本文中不进行讨论。四价或更高价态的阳离子掺杂也有研究,但高价阳离子的掺入,使得Mn3+和Mn4+的比例增加,可能会加速Mn离子的溶解。研究者还开发多离子掺杂,结合不同的过渡金属离子,或阳离子-阴离子共掺杂。这些离子可以产生协同作用,在一定程度上增强电化学性能。近年来,共混改性正极材料的研究也取得一定的进展,被证明是在电池生产过程中改善电化学性能的一种很有前景的方法。对于LiMn2O4,研究人员发现将LiMn2O4与其他正极材料(如LiCoO2、LiFePO4和Ni基正极)共混可以抑制Mn离子的溶解并提高这些材料的热稳定性,从而极大地促进了LiMn2O4正极材料的广泛应用。
Mn离子的溶解通常发生在电极表面,因此表面改性,例如包覆、表面掺杂等,是稳定正极/电解液界面(CEI)和/或在尖晶石LiMn2O4上形成人工保护层以阻止Mn溶解的另一种可控且有效的方法。非活性氧化物、氟化物或磷酸盐是常见的包覆材料,作为HF清除剂,可以有效抑制HF损伤,和/或阻碍尖晶石颗粒与电解液直接接触,从而抑制Mn的溶解,保持循环过程中的结构完整性。表面离子掺杂,如Ni、Al、Mg等,极大地增强了电极的稳定性。表面离子的掺杂,如Ni、Al和Mg,实际上大大提高了电极的稳定性。此外,许多功能电解质添加剂,如Tris(trimethylsilyl)borate、Vinylene carbonate, Succinic anhydride等被开发出来,用以消除HF和H2O等,改善固体(阳极和阴极)-电解液-界面层,进一步促进尖晶石LiMn2O4基材料的商业应用。
6.2 针对LiMnO2基正极材料
据我们所知,用以提高正交结构o-LiMnO2材料性能的方法是相对有限,相反,m-LiMnO2材料则得到了更多关注。然而,LT m-LiMnO2表现出快速的容量衰减,并在后续的循环中,遭受严重层状-尖晶石相变。随后,Bruce等人进一步报道称,在NaMnO2前驱体中,用Co或Ni等部分取代Mn,可以显著提高通过离子交换得到的LiMnO2基正极材料的循环稳定性和倍率性能。有趣的是,当Co或Ni取代量在10%左右时,离子交换得到的样品呈现出R(3–)m对称性,而不是C2/m对称性,且层状到尖晶石的转变也得到缓减。需要注意的是,软化学方法不是工业解决方案,其成本太高。相比之下,HT m-LiMnO2更具吸引力,但理想的m-LiMnO2在高温下几乎不可能存在,因此许多研究团队一直在寻找含有少量其他金属离子掺杂的高锰基m-LiMnO2材料。研究人员发现,通过用Al3+、Cr3+或Ga3+离子替代5%–10%的Mn离子,可以在高温(> 900 oC)下合成稳定的层状m-LiMnO2结构。然而,HT Al/Ga-LiMnO2表明,掺杂元素仅能减缓层状结构向尖晶石结构的转变,而不能完全避免这种转变的发生。Cr离子的掺杂则在抑制层状到尖晶石相转变方面特别有效,但循环样品仍会经历这一转变的第一阶段(5.3节中已有讨论),并显示部分Mn离子从初始八面体位点无序地位移到位于过渡金属和锂层之间的四面体位点。不幸的是,Cr离子具有高度的生物毒性。足量的Ni和/或Co掺杂(如三元、高镍等正极)确实会产生显著的差异;然而,与FMCMs相比,这些材料不再便宜,尽管它们已逐步应用于电动汽车。总之,在稳定LiMnO2材料方面已经取得一定的进展,但较为有限,仍需大量的工作。
6.3 针对Li2MnO3基正极材料
纯Li2MnO3存在严重的氧气释放、副反应和相变问题,没有展示出预期的性能,因此对Li2MnO3的改性相对较少,且效果不显著。Li2MnO3基富锂正极材料(或Li2MnO3*LiMO2,其中M = Mn、Co、Ni等)由于其基于阴阳离子参与电荷补偿,能量密度更高,尽管结构较复杂,但自提出以来越来越受到关注。有研究者认为富锂锰基材料是由C2/m Li2MnO3和LiMO2的纳米相复合而成,但也有研究者认为其是由C2/m或R(3–)m相的固溶体组成,并且双方均从不同的实验表征得到证实。事实上,二者并不矛盾,材料呈现的结构特征取决于不同的合成工艺,是固溶体还是复合相取决于畴区的大小,当畴区局限于数个原子尺度,复合相也就可看成固溶体。在富锂锰基正极材料中也出现由于类Li2MnO3组分活化而引起的O2释放。许多研究小组采用掺杂、包覆和表面处理等改性方法来降低阴离子O的活性或阻碍O2的产生,从而提高富锂锰基正极材料的电化学性能,并取得了良好的效果。
电压衰退是阻碍富锂锰基正极材料应用的重要挑战之一,因此,许多研究探讨了电压衰退的起源和机理,并采取诸多方法来解决这一问题。实际上,富锂锰基正极材料可以在4.5 V以下的低截止电压下很好地循环,而不会出现电压衰退或滞后现象,然而,在这种情况下,类Li2MnO3组分不会被激活,这是以牺牲大量容量为代价的。几乎所有发生在富锂锰基正极材料中的问题都源于类Li2MnO3组分的激活,其伴随着阴离子O的氧化还原和过渡金属离子的重排。Tarascon组很早就利用Li2RuO3作为模型,研究了不同替代元素Ti和Sn对富锂正极材料电压衰退的影响,他们通过球差电镜观测到循环后的钌基富锂材料中过渡金属会发生由过渡金属层八面体位到锂层四面体位的不可逆迁移。他们分析认为小尺寸的过渡金属如Ti4+易于发生不可逆迁移(TM层的TM迁移到Li层四面体位)导致电压衰退现象,因此可以通过向体相中引入大尺寸离子的途径来解决电压衰退问题。更多的研究人员则倾向于将富锂正极中的电压衰减与尖晶石相畴和/或立方岩盐相的形成相联系,其也涉及过渡金属离子在层内、层间的迁移。Chen等人提出,在Li离子脱出的过程中产生的氧空位和Li空位,是Mn离子迁移到Li层的原因,并带来了电压衰减。借助中子粉末衍射(ND),Mohanty等人提出,富锂正极材料中层状到尖晶石的结构演化是通过四面体阳离子中间体实现的。具体来说,首先,锂层中的锂离子从八面体扩散到四面体(LiLioct → LiLitet),接着TM层的Li从八面体位置扩散到Li层的四面体位置(LiTMoct → LiLitet);然后,TM离子通过Li层四面体位从TM层八面体位迁移到“永久”Li层八面体位(MnTmoct → MnLitet → MnLioct),导致类尖晶石相的形成和电压衰减。Wang等人观察到富锂锰基正极材料中两种不同的层状结构:R(3–)m LiMO2和C2/m Li2MO3相均逐渐转变为尖晶石结构,这与持续发生的电压衰退相对应。一些研究人员认为,尖晶石结构将进一步转变为NiO型岩盐相,因为高电压下(> 4.6 V),材料结构并不稳定。通过STEM,他们发现在层状结构和无序岩盐相之间的中间区域形成了类尖晶石相。正是尖晶石相和无序岩盐相的形成,可以解释富锂正极材料在连续循环过程中的电压衰减和容量退化。Hu等人发现,纳米尺寸的微结构缺陷,尤其是预锂化产生的大量晶界,极大地加速了O的损耗和电压衰退。他们还认为,氧释放激活低电压的Mn3+/Mn4+和Co2+/Co3+氧化还原电对,从而导致持续的电压衰退。
因此,涉及调控阴离子O,尤其是暴露在表面的O,的活性(例如降低O氧化还原活性或抑制O2释放),以及避免过渡金属离子迁移的策略,均可用以提高材料电压稳定性。表面修饰方法,如包覆、表面掺杂等,对缓减电压衰减是有效的。许多惰性氧化物、氟化物或金属磷酸盐(如Al2O3、CeO2、TiO2、AlPO4和AlF3等)等被用作包覆材料,用以保持阴离子O氧化还原产生的氧空位或保持结构完整性。碳基材料的包覆不仅可以阻止O2的释放,还可以增加电极的导电性,从而提高电极的循环稳定性和倍率性能。Meng等人则直接通过构建具有氧空位的表面,实现了对O活性的精细控制,对电极的性能产生显著影响。表面离子(如Nb、Ru等)掺杂的富锂正极材料由于表面结构稳定,在循环过程中表现出温和的阴离子O氧化还原活性,因此表现出良好的性能。许多研究小组则通过构建更稳定的尖晶石或非富锂外层,从而避免严重的氧损失。Zhu等人报道,通过选择性熔融钼酸盐辅助Li-O浸出工艺,获得具有Li离子梯度结构的过渡金属氧化物正极,其中氧损失几乎为零,并表现出非凡的电化学性能。
研究人员还采用体相掺杂的方法来调节材料电子结构以改变电子状态。例如,Li等人在富锂正极材料中掺杂多聚阴离子(BO3)3−和(BO4)5−,以降低O 2p的能级,从而减少阴离子活性。一些活性过渡金属离子被用于参与电荷补偿,以减少阴离子O的氧化还原程度。一些研究团队通过使用更多活性离子中心作为缓冲区或通过人工表面预重构,保持源于O、Mn、Ni等元素容量贡献的比例稳定,避免低电位氧化还原电对的激活,也极大地稳定了循环过程中的电压。此外,Zr、Sn、La等大尺寸离子也被用于减缓渡金属离子迁移,从而抑制材料层状向尖晶石结构的转变,从而避免严重的电压衰减。提及与电压衰退相关的结构问题时,毫无疑问,电压衰退与O3构型结构息息相关。在O3构型结构中,过渡金属离子迁移到锂层的八面体位以及层状向尖晶石结构相转变很容易发生。因此,一些研究团队探索可以避免层状-尖晶石转变的新结构。文献已经表明,O2构型LixMO2几乎不可能经历层状-尖晶石转变,因为氧离子的晶格重排需要断裂所有的Mn–O键。基于此,一些研究组通过离子交换方法成功合成多种O2构型的富锂正极材料,其电压曲线稳定,无明显电压衰退。总之,富锂锰基正极的电压衰退已经得到很好的研究和解决。
至于电压滞后,目前还停留在现象描述和定性解释的阶段,相关的研究很少,更不用说如何解决。
对于富锂正极材料的电压迟滞现象,人们提出几种解释机制,如迟缓的阴离子O氧化还原、过渡金属离子可逆迁移以及这些因素的结合。部分课题组认为,固态阴离子O电荷补偿的迟缓动力学是关键原因,类似于Li-O2电池那样,导致巨大的过电位。然而,这也带来了一些问题,如图5所示。i,GITT不能消除富锂正极中的电压差,这与Li-O2电池中的情况有所不同,如图5a和b所示;在富锂正极材料中,在高电压状态下形成的氧物种是与高价过渡金属离子配位,而不是与Li-O2电池中的过渡金属原子配位。ii,如图5c,阴离子氧化还原速率并不慢。iii,仅在阴离子O氧化还原的电压区间内(3.5~4.6 V)其电压滞后并不大,在全电压范围内才明显(图5d和e)。iv,含阴离子O氧化还原的正极材料也可表现出极小的电压滞后(图5f和g)。显然,仅考虑动力学因素是不够的。
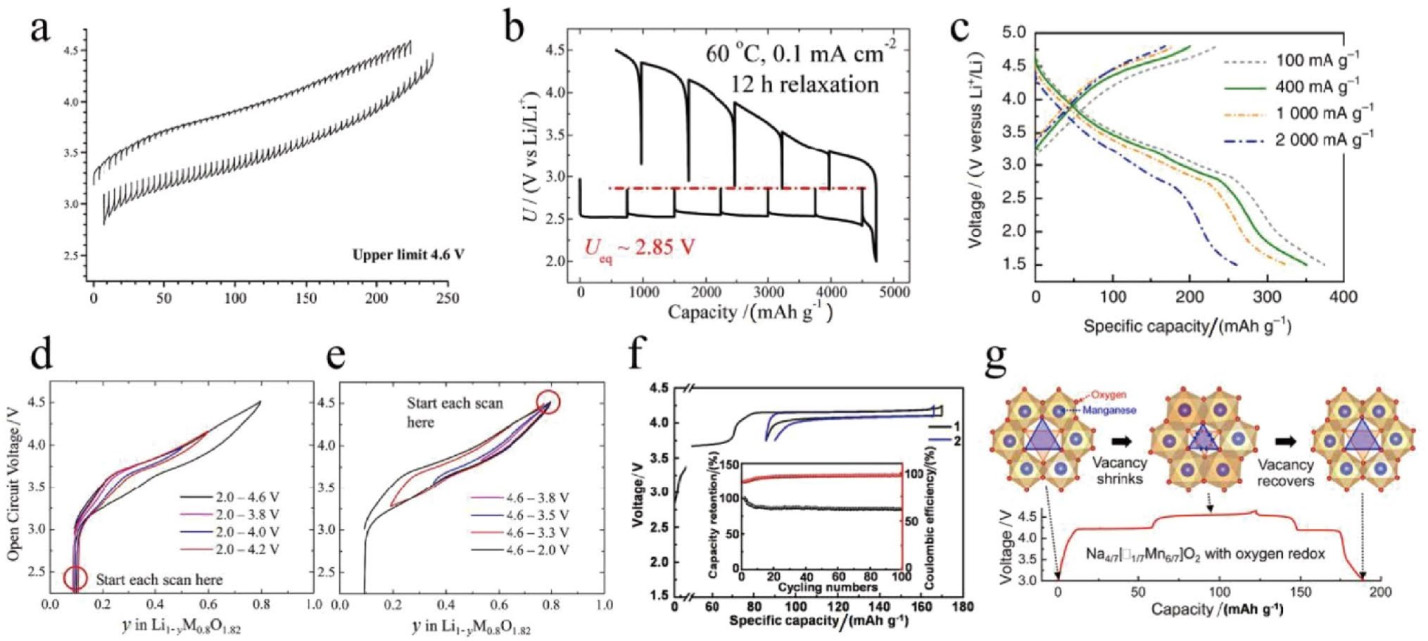
图5a 活化后第二圈富锂锰基正极材料经过GITT测试后得到的电压-容量曲线,Copyright 2014, Elsevier。b Li-O2电池的GITT曲线。Copyright 2015, Royal Society of Chemistry。c Li1.68Mn1.6O3.7F0.3的恒流充放电曲线。Copyright 2020, Nature。GITT测得的开路电压。d 从2.0 V开始扫描,充电截止电压不同;e 从4.6 V开始扫描,放电电截止电压不同。Copyright 2013, American Chemical Society。f Na0.5Ni0.25Mn0.75O2的电压曲线,插图为循环性能。Copyright 2018, Elsevier。g Na4/7[ ]1/7Mn6/7O2的充放电曲线。Copyright 2019, American Chemical Society
一些研究小组认为,过渡金属离子的部分可逆迁移使体相中O的氧化还原电对电位下降大于1 V,导致阴离子O和阳离子在循环过程中氧化还原电位的重新排序,这是通过过渡金属离子进行层间迁移,改变阴离子O的能带来说明阴离子在低电位还原的原因。这可以很好地解释第一圈的电压容量曲线,但他们没有进一步解释后续循环中的滞后现象。Croy等人也认为过渡金属离子在过渡金属层的八面体位置和Li层的亚稳态四面体位置之间的可逆迁移会影响邻近的锂离子,所以一小部分锂离子会在位点能上经历约1 V的滞后。Doublet等人统一了Li/Na离子电池中阴离子氧化还原的图像,提出O在不同的氧化还原状态下表现出不同的可逆性。随着O-O距离的缩小,狭窄的O 2p孤带分裂成σ、π、π*和σ*离散带,从而形成(O2)2−过氧化物。当氧的参与程度增加,O-O距离靠近,有阴离子O的能带进入σ*/M(d)能带反转区间,由于σ*/M(d)能带的反转,在之后的放电过程中会先发生阳离子的还原,之后才是阴离子参与,导致电压滞后,这可以看成是O二聚化导致的阴阳离子电对重新排序。
Tarascon等则发现,阳离子氧化还原路径是完全可逆的,而阴离子氧化还原则采用不同的亚稳态路径,充放电过程焓势不相等,这导致了准静态电压滞后;准静态电压滞后与非平衡熵产生的散热有关,这从热力学角度提供了很好的思路。他们还发现在阳离子无序体系中,存在配体到金属的电荷转移,中间激发态(如Ni3+/4+)构建新的非平衡态路径,是阴阳离子电对反转和电压滞后的原因。Bruce等人首先将正极材料首圈滞后行为和超晶格结构关联。他们证明TM层中Li和过渡金属离子的层内超结构决定了钠离子电池的首圈滞后。经过充分的实验表征和理论计算,他们发现超结构显著影响阴离子氧的氧化还原的行为:具有蜂窝状超晶格的Na0.75[Li0.25Mn0.75]O2电极在充电到高电位时,分子O2会在材料内部固体形成;而在带状超结构的Na0.6[Li0.2Mn0.8]O2电极中,锰离子的无序化被抑制,分子O2的形成也被减轻,阴离子O部分以稳定电子空穴参与电荷补偿,从而抑制滞后。在富锂锰基正极材料Li1.20Ni0.13Co0.13Mn0.54O2中,他们也观察到分子O2的形成。受阴离子O氧化还原的驱动,过渡金属层内原子进行层内迁移,形成可容纳分子O2的过渡金属空位团。在放电到3.75 V的较低电位,他们还发现分子O2可以被还原为O2−。基于此,他们认为首圈活化过程中,分子O2的形成以及面内过渡金属离子迁移,形成包含过渡金属空位簇的结构。在放电时,层内结构仍保持无序,Li+返回到TM层,填充空位簇中的位点形成不同于初始状态的“含锂态”,此时出现“Li6-O”的O配位环境,对应的O 2p能量是高于“TM2Li4-O”的。这导致第一次放电时的电压低于第一次充电时的电压。在第二次充电过程中,过渡金属层无序始终存在。持续的无序以及“Li6-O”氧配位导致第二次循环时氧化电位降低。此后,分子O2形成和还原产生的应变释放可以解释后续路径相关的滞后现象,这可以看成是分子O2形成,改变O的局域环境,进而改变O的能级,重新调整阴阳离子氧化还原顺序,导致滞后。
上述模型提供了富锂正极材料电压滞后的各种定性解释,但滞后行为与阴离子O氧化还原或过渡金属迁移的定量关联仍然是一个不明确且具有挑战性的问题。此外,当我们谈论Li2MnO3组分活化过程中,过渡金属离子重排时,仍有较多问题,有太多细节缺失。i)Li2MnO3中发生的阴离子氧化还原反应与富锂锰基正极材料中发生的阴离子氧化还原反应大有不同,其他活性过渡金属离子,如Ni、Co等的作用是什么;并非所有的Li2MnO3都没有电化学性能或阴离子氧化还原可逆性。ii)活化过程中离子的重排程度如何以及形式怎样?iii)在不同的充放电状态下,离子具体占据哪些位点,这些位点是否是热力学稳定的,以及如何准确表征?iv)在随后的循环过程中,离子是如何迁移的?v)在充放电过程中,从颗粒到颗粒、表面到体相,电极的组成和结构是如何变化?vi)在活化后的充放电循环中,为什么活化结构不能表现出完全的可逆性,GITT也不能完全消除电压差?仍需更多工作来解释这个棘手且至关重要的问题。
7. 新材料、新方法
过去几十年,研究人员在FMCMs方面做出了大量努力并取得了显著进展。令人鼓舞的是,最近一些团队正开发出一些新颖的FMCMs和新的改性方法,并取得了重大突破。
7.1 阳离子完全无序和部分有序氧化物正极
Ceder课题组首次将渗流理论引入LIBs领域,合成了阳离子无序岩盐氧化物正极,并对其进行深入研究。他们发现,Li1.211Mo0.467Cr0.3O2(LMCO)的层状结构特征在循环一次后开始丧失,在10次循环后基本完全消失,如图6a所示。循环后样品的STEM图像显示出明显的阳离子无序原子排列。事实上,几十年来,阳离子无序一直被认为是不利于锂离子传输的,在这种结构中没有1D、2D或3D锂离子通道。然而在阳离子无序之后,LMCO电极仍然释放出非常高的可逆容量(> 265 mAh g−1),如图6b所示,这是有违直觉的。他们提出Li离子在阳离子无序岩盐相中的扩散可以通过八面体-四面体-八面体跳变(即o-t-o扩散)来实现。在中间体四面体位的Li是Li扩散的激活态。考虑到激活态与过渡金属离子共面的数量,他们给出了在Li-TM氧化物中三种可能的扩散通道:0-TM、1-TM和2-TM通道,如图6c所示。激活态的能量反映了锂离子的迁移势垒,主要是由激活态的锂离子与其共面元素之间的静电斥力所决定,因此取决于共面元素的价态、激活态锂离子与共面元素之间的驰豫空间。0-TM通道和1-TM通道均支持o-t-o机制,但当有两个以上共面的过渡金属离子存在时,激活态锂离子会面临强烈的静电排斥。所幸的是,与1-TM通道相比,0-TM通道表现出相对较低的势垒,这表明锂离子在无序LMCO电极中的迁移可能较为容易。他们进一步证明了阳离子无序岩盐正极材料的可行性:当LixTM2−xO2中的x超过~1.09(渗流阈值)时,可以打开0-TM通道网络,如图6d所示。增加锂离子的浓度,使渗流0-TM网络中增加了更多的0-TM通道,进而增强渗流网络的连通性。因此,锂离子可以通过阳离子无序岩盐相材料中的0-TM通道跳跃,实现锂离子的可逆脱嵌。这种材料拓宽了研究人员对正极材料的想象空间,其中,阳离子无序全锰基正极材料近年来取得重要的进展。阳离子无序Li2MnO3(Li[Li1/ 3Mn2/3]O2)或基于Li2MnO3的FMCMs满足临界条件(Li > 1.09),已被多个课题组成功合成,具有较高的电化学活性。有趣的是,Yabuuchi等人开发出阳离子无序结构的LiMnO2,并表现出良好的电化学活性,但根据渗流理论,其锂含量未达到渗流阈值。他们认为锂过量并不是从阳离子无序结构中脱嵌锂离子的必要条件;相反,缩短颗粒内的迁移长度也可以获得优异的电化学性能。
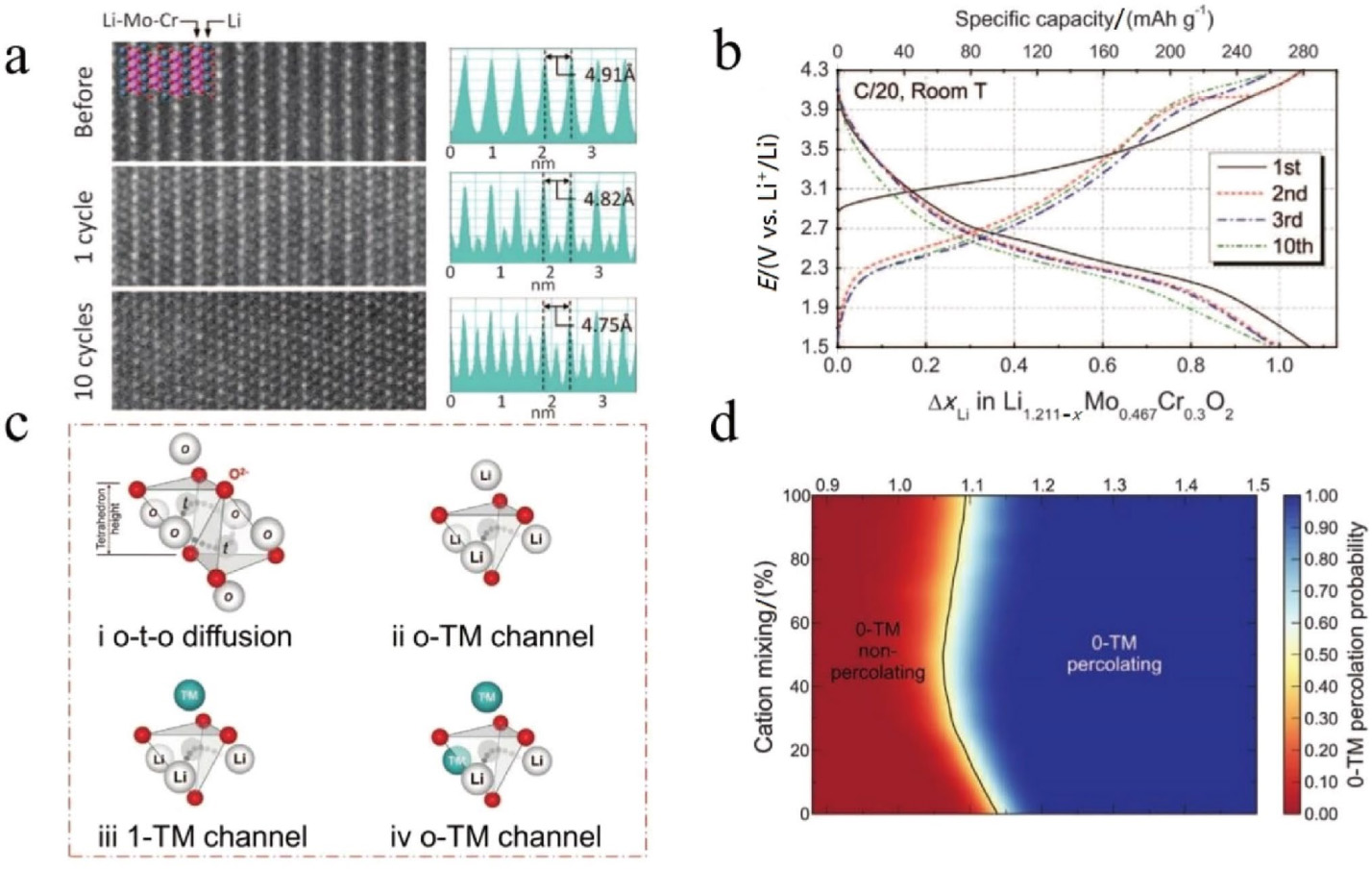
图6 a 不同循环次数后下Li1.211Mo0.467Cr0.3O2 (LMCO)正极极的STEM图像。b LMCO的电压曲线图。c 在类岩盐相Li-TM氧化物中o-t-o 迁移的可能环境。d 找到0-TM通道渗透网络的计算概率。Copyright 2014, American Association for the Advancement of Science
此外,一些研究人员利用基于非常规Mn离子和O离子的多电子氧化还原反应,实现了超高容量FMCMs。Pralong团队首次报道了一种阳离子无序的“Li4Mn2O5”正极材料,该材料通过直接机械化学方法制备得到。通过磁性测试,他们证实这种材料通过Mn3+/Mn4+/Mn5+和少量阴离子O的氧化还原,可提供高达355 mAh g−1的非凡放电比容量。随后深入的密度泛函理论(DFT)计算结果显示,脱锂过程通过一个复杂的三步反应路径进行,涉及阳离子和阴离子氧化还原反应的相互作用。在充电阶段初期,主要是Mn离子的氧化:Mn3+ → Mn4+(LixMn2O5,4 > x > 2);之后阴离子O参与电荷补偿:O2− → O1−(2 > x > 1);最后,金属离子进一步氧化,Mn4+ → Mn5+(1 > x > 0)。与八面体配位的Mn4+相比,四面体配位的Mn4+在能量上更有利于氧化成Mn5+。因此,他们认为在Mn2O5的最终氧化过程中,需要Mn离子迁移到四面体位点,从而使Mn4+氧化为Mn5+。基于Mn2+/Mn4+和少量的阴离子氧化还原,Ceder团队也实现了接近1 000 Wh kg−1的能量密度。通过高价阳离子(Nb5+、Ti4+)对Mn离子以及F−对O2−的部分取代,他们成功地合成了含Mn2+的富锂正极材料。此类材料增加了正极材料的多样性和可设计性。然而,在阳离子完全无序的正极材料中,材料倍率性能和循环稳定性都不够好。Ceder等人进一步采用,将大量锂过量与部分类尖晶石阳离子有序结构相结合的新策略,获得了大于1 100 Wh kg−1的比能且放电电流密度可达20 A g−1,改变了在无序材料中,锂离子扩散缓慢的固有观念,为锂离子正极材料实现致密和超快储能提供模型。
7.2 调控原子排列有序度和结构对称性
越来越多的研究人员开始关注正极材料中过渡金属层内离子的排列和分布,而不仅仅是宏观晶体结构。课题组等通过过渡金属层内Li、Mn无序化,如图7a,合成了一种R(3–)m对称性的全锰基Li0.700Li0.222Mn0.756O2富锂正极,该材料具有高容量、优异的倍率性能和良好的循环稳定性。由于特殊的原子排列和更高的结构对称性,制备的材料中Mn3+离子处于低自旋状态,从而缓减了Mn离子的Jahn-Teller畸变。阴离子O的反应活性和锂离子扩散也得到调节,展现出更为温和的阴离子氧化还原活性和快速的二维锂离子通道。进一步研究发现,在层内Li、Ru无序的Li2RuO3中,阴离子O的氧化还原反应则通过O-Ru-O伸缩进行结构响应,而不会发生O-O二聚化。Cho等人也探索了富锂层状氧化物中持续的阴离子氧化还原化学、过渡金属离子迁移与锂过量局域化的关系。计算结果表明,对于锂过量局域化的富锂正极材料(L-LMR,图7d),其在费米能级附近因“Li–O–Li”构型诱导产生的O p带中心,相较于锂过量非局域化的富锂正极材料(D-LMR,图7g),发生上移,如图7e和h所示。因此,与L-LMR相比,D-LMR表现出更温和、可逆的阴离子氧化还原活性,以及更稳定的结构完整性,因此性能优异,如图7f和i所示。

图7 a R(3–)m Li0.700Li0.222Mn0.756O2沿[1010](R(3–)m)方向观测得到的HAADF-STEM。b 计算得到的C2/m Li[Li1/3Mn2/3]O2和R(3–)m Li0.75Li0.25Mn0.75O2部分Mn和O的态密度图(pDOS)。c R(3–)m Li0.700Li0.222Mn0.756O2的充放电曲线。Copyright 2020, Wiley-VCH。d L-LMR和g D-LMR[100]单斜方向的HAADF-STEM。e 过量锂局域化体系和 h 过量锂非局域化体系的O 2p轨道和TM 3d轨道的pDOS图。f, i L-LMR和D-LMR的首圈和第2圈电压曲线。Copyright 2020 Wiley-VCH
对于尖晶石LiMn2O4基的FMCMs, Amine等人研究表明,理想LiMn2O4在充放电过程中发生了严重的不可逆相变,产生意想之外的相,如Mn3O4、Li4Mn5O12、过锂化Li2Mn2O4等,并在颗粒表面产生裂纹,带来糟糕的表面稳定性及快速的Mn离子溶解。反过来,Mn溶解结合Jahn-Teller畸变,进一步加速不可逆相变,导致容量恶化加剧。因此,他们开发了一种具有Li、Mn无序排列和表面重构的富锂Li1.09Mn1.91O4正极材料,可以有效抑制Mn离子的溶解和不可逆相变,即使经过25次循环,在XRD图中也几乎没有检测到意想之外的相。Pan等进一步提出,阳离子无序可以从本质上抑制Mn3+-O6八面体的协同Jahn-Teller畸变,避免Mn3+O6八面体沿一个方向伸长,抑制尖晶石结构中Mn3+的歧化。通过这种策略,他们在微米尺度LiMn2O4基材料中实现双倍容量的良好稳定循环。
至此,毫无疑问,原子排列有序度可以显著影响材料的本征性质及电化学性能。可通过完全无序或部分无序的原子排列来调控材料晶体结构,对正极材料电化学性能的稳定性和可逆性具有重要影响。更进一步,在过渡金属层中Li离子和/或过渡金属离子更为均匀的分布,如过渡金属层内离子无序,或超晶格的概念(过渡金属层中Li、TM周期性排列),也可以有效地影响材料的性能。通过过渡金属层内离子无序化控制正极材料的性能,在锂离子电池和钠离子电池领域中均有报道,而超晶格控制正极材料性能的报道主要集中在钠离子电池领域。其中,部分超晶格是由不同的过渡金属离子(TM/TM’)组成,部分超晶格是由不同比例的Li、M、TM离子组成。有趣的是,空位、过渡金属离子也可以在过渡金属层中形成超晶格或无序排列,并对正极材料的性能产生显著影响。在最新的文献中,就有设计和合成具有不同Li/V/TM排列的FMCMs,提供额外的可用Li存储位点和高度稳定的可逆阴离子氧化还原,极大地促进了FMCMs的发展。
结构对称性是影响正极性能的另一个重要因素。几乎所有具有R(3–)m对称的传统层状LiMO2氧化物正极都表现出比C2/m LiMnO2更好的电化学性能。由C2/m Li2MnO3和R(3–)m LiMO2构成的富锂锰基正极材料,也比纯C2/m Li2MnO3或C2/m Li2MnO3* C2/m LiMnO2正极材料表现出更高的可逆性和稳定性。当然,活性过渡金属离子,如Ni、Co等的贡献不能排除。有趣的是,Chen等通过理论计算预测,一旦LiMnO2具有R(3–)m对称结构,低自旋态Mn3+可能存在,则可能不存在明显的Jahn-Teller畸变。事实上,低自旋Mn3+离子确实能存在于层内Li/Mn无序化的R(3–)m Li0.700Li0.222Mn0.756O2材料中,并对这种FMCMs的电化学性能产生有益的帮助。近年来,Xia等人合成了一种尖晶石和层状结构域交织的LiMnO2正极。其中,Mn-dz2轨道在两畴曲界面处垂直取向,形成有序的界面轨道,抑制了协同的Jahn-Teller畸变和Mn离子的溶解,使得材料具有优异的循环性能。操空Mn离子的电子结构,特别是自旋态的工作,值得更多的关注。
7.3 调控O亚晶格
层状LiMnO2或富锂Li2MnO3基正极中发生的Mn离子迁移、层状到尖晶石相变,无疑与O3构型息息有关。O3构型与尖晶石结构具有相似的O面心立方堆积,因此,在O3构型锰基层状氧化物正极材料中较为容易实现层状到尖晶石的转变。一旦Mn离子迁移到中间Li层四面体位,就很有可能迁移到邻近的锂层八面体位点,完成层状到类尖晶石状的转变,这是因为在锂层脱锂过程中形成的八面体空位仅与MnO6八面体共边。因此,通过制备具有不同氧亚晶格的FMCMs,例如隧道型或O2构型FMCMs,可以有效避免这种转变。O2构型FMCMs是由少数研究组较早提出和合成的,由于没有层状到尖晶石的相变,也就没有明显的电压衰退。事实上,对于O2构型Li-TM氧化物而言,由于共面阳离子之间存在较大的静电排斥作用,可以预计其不利于过渡金属离子从中间位点迁移到相邻的Li位点。因此,在O2构型正极材料中,层状到类尖晶石相变在室温下几乎是不可能发生的。目前已经有部分团队成功合成一系列性能优异且无明显电压衰减的O2构型得富锂正极材料。然而,目前是通过离子交换法控制层状氧化物正极材料中的氧亚晶格。离子交换法,暂时还不是一种工业化的方法。直接原位电化学方法合成具有不同氧亚晶格的FMCMs或其他新方法均值得尝试。
7.4 界面或表面氧化还原
在插层式正极材料中,通常会发生相对缓慢的体相扩散控制的反应,导致其有限倍率性能。因此,一些研究团队将重点放在界面或表面氧化还原反应上,以同时实现高容量和高倍率性能。Kang等人证明在无锂金属单氧化物(MO)的表面经过纳米级LiF装饰后,可以转化为高容量和高倍率的正极材料。该材料展现出平滑的电压特性曲线、更高的容量和较为出色的倍率性能。这主要归因于基于Mn离子氧化还原的表面转化反应,与经典的Li离子嵌入反应不同。课题组研究发现,可通过PF6−在过渡金属氧化物(例如Mn3O4)表面进行可逆吸/脱附,实现电解质阴离子氧化还原吸附式赝电容效应。其中,Mn3O4与PF6−之间可通过Mn–F键进行电荷转移,F离子作为氧化还原中心,为高容量和高功率储能器件提供了更大的设计自由度。此外,这类材料还可以避免由于体相中锂离子嵌入/脱出引起的渐进相变和材料结构崩溃导致的电压曲线连续变化和糟糕的稳定性等问题。
7.5 熵稳定策略
熵稳定策略可能是提高FMCMs电化学性能的一种很有前景的方法。实际上,熵稳定策略在传统合金领域早已得到广泛应用,并在能量转换和存储方面取得巨大进展。在我们早期的研究中,从有序的Li2MnO3到过渡金属层内阳离子无序的Li-Mn-O正极材料,再到常规的富锂锰基正极材料,材料的循环稳定性随着层内构型熵的增加而提高。近年来已经报道了一些高熵电极材料,但主要集中在钠离子电池正极材料、锂离子电池氧化物负极和阳离子无序正极材料方面。这些新型氧化物体系也被称为高熵氧化物(HEO),几年前就被首次提出。Breitung等人针对几种TM-HEO的电化学性能进行了一些新的研究,如储存容量和循环稳定性。结果表明,熵的稳定效应显著地提高了循环稳定性,并为HEO的储存容量的保持提供了重要支撑。此外,还观察到HEO的电化学行为取决于每种金属阳离子的存在,为通过简单改变元素组成,调节材料的电化学性质打开大门。Hu等人报道了钠离子正极高熵化学的新概念。层状的O3构型NaNi0.12Cu0.12Mg0.12Fe0.15Co0.15Mn0.1Ti0.1Sn0.1Sb0.04O2正极表现出良好的循环稳定性和出色的倍率性能,这归因于在Na+(脱出)嵌入过程中,多组分过渡金属对局部相互作用变化的适应。他们进一步发现,层间距扩大的多组分TMO2层有助于加强层状氧化物整个骨架结构,从而大大提高电化学性能和热稳定性。在锂离子电池正极材料领域,目前仅有少数高熵阳离子无序正极材料被报道。多元素的引入可以带来协同效应,有利于循环稳定性和热安全性,值得关注。
7.6 搭配固态电解质
有毒、易挥发和易燃的有机电解液在商业锂离子电池中已被广泛应用,因此存在一些潜在的安全问题,包括电解液泄漏、火灾和爆炸等。固态电解质是一颗冉冉升起的新星,因其高安全性、宽工作温区和便于回收而受到越来越多的关注。此外,固态电解质允许使用锂金属作为负极,这反过来又增加电池电压,从而提高电池的能量密度。当前固态电解质系统可以大致分为三类:(1)无机电解质(包含氧化物基、硫化物基固态电解质等),(2)固体聚合物电解质,和(3)复合电解质。FMCMs,尤其是高电压、高容量的FMCMs,搭配固态电解质将产生重要影响:除了高安全性和高能量密度的优点外,共同的问题——Mn溶解在理论上可以通过固态电解质来解决,这将成为未来最重要的发展方向之一。
经过近一个世纪的探索和发展,固态电解质已经取得了一定成就,但在走向商业化的道路上仍存在一些不足和挑战,亟待解决。由于电池的性能高度依赖于电解质中锂离子的扩散性能和界面相容性,因此固体电解质需要具有高的离子电导率、可忽略的电子电导率和良好的渗透性;此外,考虑到实际应用,固态电解质还应具有良好的力学性能。几乎所有的无机氧化物电解质都面临电极-电解质界面接触不良的问题,在循环过程中界面稳定性也较差,导致界面阻抗快速增加,容量快速下降。因此,氧化物固态电解质往往需要加入一些聚合物组分,与微量离子液体或高性能锂盐和电解液混合,或使用辅助原位聚合制造准固体电池,以保留一些安全优势并改善电解质-电极界面接触。因此,氧化物固体电解质通常需要添加一些聚合物组分,与微量离子液体/高性能锂盐和电解质混合,或者使用辅助的原位聚合来制备准固体电池,以保留一些安全优势并改善电解质-电极界面接触。与氧化物电解质相比,硫化物基电解质具有更高的离子电导率,室温下可达10−3 S cm−1,是固态电池理想的电解质材料。但其稳定性(电化学稳定性和界面稳定性)仍有待解决。此外,大规模应用的主要问题是:硫化物对空气的脆弱性,任何处理都必须在惰性气氛中进行。此外,无机电解质中电极和电解质的合成过程与标准锂离子电池技术不同。可用的粘结剂和溶剂种类较少,且降低固态电解质厚度也较为困难。相比之下,固态聚合物电解质,尤其是原位聚合固态聚合物电解质,是另一种很有前途的固体电解质,具有安全性好、易加工成膜、界面接触好等优点。同时,它还可以防止锂枝晶问题,这是近年来备受关注的。然而,许多挑战仍然存在。其中,开发具有高离子电导率和宽电化学稳定窗口的聚合物电解质尤为重要。与纯固体聚合物电解质和无机电解质相比,复合电解质具有以下优点: (1)可结合高机械强度和高离子电导率,活性无机填料还可提供自由移动的锂离子;(2)形成有序或无序的无机材料-聚合物界面结构,为锂离子迁移提供了输运通道,带来更高的离子迁移数;(3)无机材料的存在还可以吸附微量水和其他微量杂质,使复合电解质在电化学环境中更加稳定,拓宽了电解质的电化学窗口。然而,无机材料与有机聚合物电解质之间存在相界,在长时间的充放电循环中发生相分离,导致放电容量急剧下降。无机填料在聚合物电解质中不易分散,纳米级无机颗粒容易团聚,导致电池循环稳定性下降。此外,复合电解质的制备工艺和条件更为复杂,成本也较高。
搭载固态电解质的FMCMs电池的开发是一个系统工程,需要进一步探索固态电解质内在机制、开发新型高性能固态电解质、构建和调控电极/电解质界面以及应用先进表征技术进行精确表征。还有需要开展更多的工作。
06 重要结论
FMCMs具有资源丰富、生物毒性低、晶体结构可控和多变价Mn离子氧化还原电对等优点,在下一代锂离子电池中具有巨大的竞争优势和潜力。然而,FMCMs仍然面临诸多具有挑战性问题,这些问题不容忽视,主要归因于锰离子的电子结构和配位结构,以及电极材料的晶体结构。基于上述研究进展及课题组的实验分析,我们提出以下结论和展望,旨在为构建更好的FMCMs提供一些有吸引力的策略和研究方向,如图8所示,以下是更详细的说明。

图8 从Mn离子电子结构、配位结构以及材料的晶体结构视角出发,构建更好FMCMs的可能策略。
(1)大多数锂锰氧化物正极中的锰离子具有6个氧配体,形成MnO6八面体。然而,一旦存在足够多的高自旋态Mn3+离子,就会发生协同Jahn-Teller畸变,严重影响SEI和电极结构的稳定性,从而大大降低材料的循环稳定性。因此:
i. 避免Mn3+作为Mn-氧化还原的中间体是最为直接的想法,但这并不是一个实用的建议,这使得难以实现更高容量的释放;因此,一些研究团队引入其他氧化还原中心来降低Mn3+/4+参与氧化还原的程度,例如在富镍正极材料中引入Ni2+/4+氧化还原电对或在富锂锰基正极材料中引入阴离子O的氧化还原。尽管这些方法可以有效减少Mn3+/4+的氧化还原反应,但也带来了其他令人困扰的问题。
ii. 抑制高自旋态Mn3+作为氧化还原的中间体,已有研究表明,可通过结构对称性调控来实现。
iii. 通过破坏(层状/尖晶石)锂锰氧化物中Mn3+离子的周期性排列,可以缓解协同Jahn-Teller畸变。例如,构建无序或部分无序的材料,包含:过渡金属层内,空位/Li/TM无序排列;构建除蜂巢状超结构以外的他超结构材料,控制过量锂;高熵氧化物正极材料(高层内构型熵正极,或是阳离子无序高熵正极)的合成;或调控界面轨道有序度。
iv. 通过配体取代,打破MnO6配位结构,如F或S取代O离子,或直接构建MnY8或MnY4(这里Y代表阴离子)配位结构。
(2)具有特定结构的FMCMs采取对应的解决方案:
i. 对于尖晶石LiMn2O4,掺杂、包覆和电解质添加剂通常是有效的;尖晶石LiMn2O4正极研究的关键或未来发展方向可能是通过多电子,如Mn2+/3+/4+氧化还原反应来提高其可逆容量,同时应认真考虑锂过量策略和/或Li、Mn有序度。
ii. 对于O3构型层状锂锰氧化物,除了传统的修饰外,氧亚晶格工程可能是一种有吸引力的方法。实际上,O2构型锂锰氧化物表现出优越的电化学性能,可避免持续的层状到尖晶石的相变,但其合成方法有待进一步探索。
iii. 对于富锂-锂锰氧化物,寻找具有不同层内超晶格的富锂-锂锰氧化物是一个有前景的方向。这可以实现对阴离子氧化还原活性和晶体结构稳定性之间的精妙平衡。在研究中需要关注Li/空位/M/TM的层内排列,这可能有助于避免严重的氧析出和过渡金属离子迁移。此外,还需要深入研究电压滞后的成因。
(3)赝电容兼具超级电容器和电池的优点,可在FMCMs中发挥重要作用。插入式赝电容行为在FMCMs中不易实现,但表面/界面氧化还原赝电容是可以实现的,这为提高正极材料的能量密度和倍率性能提供了一种新的可能策略。
综上所述,在未来提升FMCMs性能方面,存在着诸多机遇和挑战,仍有大量工作需要完成。在不久的将来,我们期望可以看到FMCMs与其他层状氧化物正极一样,快速发展和并逐步商业化。
07 主要作者简介

宋进(第一作者):北京大学博士研究生。聚焦锂离子电池正极材料设计合成,在Adv. Mater.、J. Am. Chem. Soc.、Energy Environ. Sci.、Nature Commun.等国际知名期刊发表SCI论文15篇,其中以第一作者在Adv. Mater.上发表研究性论文两篇,在Electrochemical Energy Reviews上发表综述论文一篇。

夏定国(通信作者):北京大学教授,博士生导师,承担或完成了包括国家重大研究计划、国家自然科学基金重点课题及北京市自然科学基金重点课题;在锂离子电池材料、燃料电池催化剂及材料制备等领域完成国家与省部级课题10余项;获国家发明专利20多项;获得国家与省部级科学技术奖励3项。出版学术专著2部;获得国家科技进步二等奖一项,北京市科学技术一等奖一项(基础类)。以通信作者身份在Nature Commun., Energy Environ. Sci., J. Am. Chem. Soc., Adv. Mater., Angew. Chem. Int. Ed., Nano Lett. 等学术刊物上发表研究论文150余篇。
推荐阅读
1. 吴忠帅研究员最新EER综述|二维介孔材料应用于能源存储和转换:现状、合成与挑战
2. 昆士兰大学王连洲教授课题组最新EER综述 | 富锂正极材料激活过程的调控与机理
3. 美国俄勒冈州立大学纪秀磊教授课题组最新EER综述|用于水性单价离子电池的普鲁士蓝类似物正极材料
4. 天津大学Michael D. Guiver/尹燕课题组最新EER综述|燃料电池碱性稳定阴离子交换膜:脂环族季铵基阴离子导体
关于我们

Electrochemical Energy Reviews (《电化学能源评论(英文)》,简称EER),该期刊旨在及时反映国际电化学能源转换与存储领域最新研究进展。EER是全球首本专注于电化学能源的英文综述类期刊。EER覆盖电化学能源转换与存储所有学科,包括燃料电池、锂电池、金属离子电池、金属-空气电池、超级电容器、制氢-储氢、CO2转换等。EER为季刊,每年3月、6月、9月以及12月出版。创刊号在2018年3月正式出版。
2018年6月,经过激烈角逐(87选20),EER成功入选由中国科协、财政部、教育部、国家新闻出版署、中国科学院、中国工程院等六部门联合实施的中国科技期刊国际影响力提升计划D类项目,进入新刊国家队阵列。
EER于2020年8月被SCIE收录;2021年6月,被EI和Scopus同时收录;2022年5月,被CSCD收录;2022年6月,入选《科技期刊世界影响力指数(WJCI)报告》2021版;2023年6月发布的爱思唯尔CiteScore 为42.8,3个学科(材料科学、化学工程、电化学)排名蝉联第一;2023年6月发布的JCR影响因子为31.3, 在全球电化学领域排名持续第一。目前文章篇均下载量超过5 400次。
欢迎关注和投稿
期刊执行严格的同行评议,提供英文润色、图片精修、封面图片设计等服务。欢迎关注和投稿。
联系我们

长按/扫描关注EER微信公众号
E-mail: eer@oa.shu.edu.cn
SpringerWeb: https://www.springer.com/journal/41918
ShuWeb: http://www.eer.shu.edu.cn
WeChat: ElectrochemicalEnergyReviews
原稿:作者提供
编辑:全海芹
初审:陈昕伊、全海芹
复审:何晓燕
终审:刘志强
https://wap.sciencenet.cn/blog-3390413-1414408.html
上一篇:热烈祝贺EER入选《科技期刊世界影响力指数(WJCI)报告》2023年版!
下一篇:悉尼科技大学汪国秀教授最新EER综述|面向可持续能源存储的高能室温钠硫/硒电池