博文
选哪个lncRNA的哪段设计引物?RT-qPCR验证,我要百发百中!
|||
本文转载自嘉因微信公众号,已获得授权。查看最新文章,敬请关注嘉因,微信ID:rainbow-genome
作者:小丫 来源:嘉因
前几年是lncRNA测序时代,最近进入了后lncRNA时代,人们都在研究lncRNA的功能和调控机制。
去年4月,小丫写了这篇帖子,得到的评价是“看透了lncRNA验证,但描述得不够清楚”。最近又遇到有人问这个问题,就把这帖子拿出来,重新编辑,力求逻辑更顺畅,把脑中所想传达给更多的人。
问题的提出
lncRNA-seq获得了大量差异表达lncRNA,要做RT-qPCR验证。
选哪个lncRNA?选变化倍数大的?表达量高的?
选lncRNA的哪段设计引物?跟其它基因有重叠怎么办?
总体原则
1. 用跟测序同一批次的RNA样品做RT-qPCR;
2. 按lncRNA类型排序,优先选择lincRNA(基因间的lncRNA),和位于其他基因intron区的lncRNA;与其他基因exon有重叠的lncRNA,可选择其中无重叠的区段;
3. 按表达丰度TPM排序,优先选择前50%的lncRNA;按变化倍数排序,优先选择前50%的lncRNA;
注:具体限制多少TPM或变化倍数,由整体测序深度和差异表达基因的数量决定,保留排在前面的一半左右,自己斟酌;
4. 一定在测序结果里看到明显变化的峰的位置设计RT-qPCR引物;
为什么遵循以上原则?下面逐个解析。
解决方案
一、Sample准备
RT-qPCR验证sequencing结果,是用一个技术平台验证另一个技术平台的结果,所以,除了所使用的技术平台不同以外,其他条件都要尽量相同,其中最重要的就是样品一定要相同。
测序和RT-qPCR,要求来自同一管RNA。如果要做RT-qPCR时需要再次提取RNA,那么退而求其次,要来自跟测序样品同一批次的细胞或同一块组织。
具体可以这样操作,分两种情况:
如果送RNA到测序公司。自己提取RNA,一半RNA送公司测序,保留另一半RNA,冻存于-80,最好存液氮,或者反转录后存cDNA。
如果送细胞或组织到测序公司。由测序公司提取RNA,要求测序公司将剩余的RNA返还。
测序分析结果回来了,用跟测序同一批次的RNA样品做RT-qPCR。
如果用来做RT-qPCR的样品跟测序不是同一批次,那么你验证的就是两层面的叠加:技术平台间的差异 + 生物学重复间的差异。如果结果不一致,就搞不清楚是技术造成的差异,还是生物学重复的不一致。
二、不能选哪种lncRNA
先说什么样的lncRNA不能选来做qPCR验证。
用链特异性Strand-specific技术建库的lncRNA-seq,能够区分来自正义链和反义链的转录本,而普通的RT-qPCR无法区分来自不同链的RNA。
因此,不能选与其他基因exon重叠的lncRNA,否则,RT-qPCR结果就混合了两个基因的表达量。
最好选择基因间的lncRNA,即lincRNA,跟其他基因没有重叠。
其次选择位于其他基因intron区域的lncRNA。
再次,如果lncRNA的一部分与其他基因的exon重叠,另一部分不重叠,就选不重叠的那部分。
三、选哪个lncRNA
表达量高的优于表达量低的,更容易做RT-qPCR,Ct值更低,结果更可靠;另外,表达量高的基因,其差异表达筛选时的准确性更高。
表达量用丰度TPM来衡量,点击链接查看详情:
由于测序和RT-qPCR原理不同,所以sequencing和RT-qPCR计算出来的变化倍数不可比,只要变化趋势相同就算结果一致。
四、选哪段
到IGV里查看wig文件,哪个区段峰高,并且在组间变化明显,就选哪段设计PCR引物。
lncRNA某些区域可能看不到read,这可能是以下原因导致的:1. 基因有不同isoform,在你的样品里可能只表达其中一种isoform;2. PCR或测序偏好;3. 基因结构注释有问题。
RNA-seq计算lncRNA表达量和筛选差异基因时,用的是lncRNA全长read counts的总和,不代表每个区段都有表达或表达差异。因此,选择高峰所在的区段做qPCR,能保证万无一失。
举例子
在IGV里面打开wig文件,在IGV里逐个搜索满足上述原则的lncRNA,查看read分布。
栗子一
下图这个lncRNA位于基因间区,有不同的isoform,其中一些exon的read counts较多,如图中三个较高的蓝色峰,且在两个样品间有明显的变化,这三个峰所在的位置就适合设计引物。
怎样拿到序列呢?
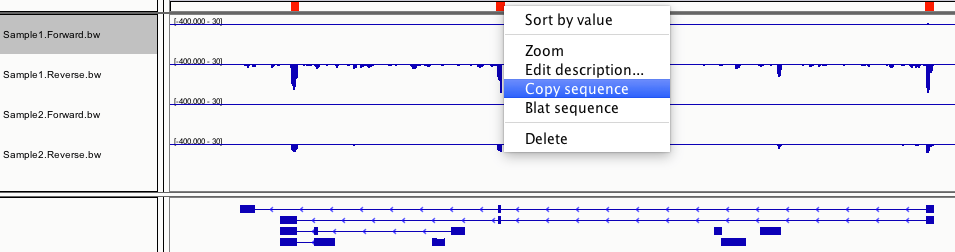
点击IGV里的这个小图标 ,然后在你感兴趣的区段左边点一下鼠标,右边点一下鼠标,就出现红色的那一段了,在红色区域点右键,copy sequence,粘贴到你的引物设计软件里,就OK啦!
,然后在你感兴趣的区段左边点一下鼠标,右边点一下鼠标,就出现红色的那一段了,在红色区域点右键,copy sequence,粘贴到你的引物设计软件里,就OK啦!
栗子二
对于链特异性建库的lncRNA-seq数据,每个sample有Forward和Reverse两个track,分别代表正义链和反义链,你能看到两条链上转录出来的不同的转录本。
下图这个lncRNA位于一个蛋白编码基因的antisense(箭头标明转录方向),虽然gene结构注释认为lncRNA位于内含子区域,但其下游仍有较多的read分布,可能是基因结构注释的问题。
我标出红色的区域跟另一个基因的exon没有重叠。
这里的四个track,分别是第一个sample的Forward和Reverse,和第二个sample的Forward和Reverse。两个sample的Reverse间有明显的差异,可用来设计引物。

最后,用哪个软件设计引物?
推荐primer3,http://primer3.ut.ee,简单高效。
Product Size Ranges限制为80-150,其余全部默认。我的经验是它设计出来的第一对引物就好用。
下面的帖子可能对你有帮助:
https://wap.sciencenet.cn/blog-3372875-1090311.html
上一篇:史上最全ncRNA数据库:RNAcentral | 最权威的EMBL-EBI出品
下一篇:全世界的miRNA分析工具都在这里