博文
GCTA PCA分析以及软件安装教程
||
软件介绍系列
1. GCTA介绍
在群体遗传中,GCTA中做PCA非常方便, 下面介绍一下GCTA的安装方法.
2. 安装命令
使用conda自动安装
conda install -c biobuilds gcta
手动安装
3. 安装成功测试
这里, 应该键入gcta64, 而不是gcta
(base) [dengfei@localhost bin]$ gcta64 ******************************************************************* * Genome-wide Complex Trait Analysis (GCTA) * version 1.26.0 * (C) 2010-2016, The University of Queensland * MIT License * Please report bugs to: Jian Yang <jian.yang@uq.edu.au> ******************************************************************* Analysis started: Wed Apr 24 14:07:43 2019 Options: Error: no analysis has been launched by the option(s). Analysis finished: Wed Apr 24 14:07:43 2019 Computational time: 0:0:0
显示上面信息, 表明软件安装成功.
4. 功能介绍

5. 参数说明
5.1 输入输出文件
输入文件:
--bfile test: 类似plink的参数格式. 支持binary文件(test.fam,test.bim,test.bed)--dosage-mach test.mldose test.mlinfo支持其它数据格式
输出文件:
--out result: 类似plink的--out参数, 定义输出文件名
5.2 数据清洗
ID保留和删除
如果不写, 默认全部使用
--keep test.indi.list定义分析个体列表--remove test.indi.list删除个体列表
选择SNP
--chr 1:选择染色体--autosome 选择所有SNP
6. 构建G矩阵
--make-grm 会生成三个文件:
如何你想在R中读取二进制文件, 可以使用如下代码:
ReadGRMBin=function(prefix,AllN=F,size=4){
sum_i=function(i){return(sum(1:i))}
BinFileName=paste(prefix,".grm.bin",sep="")
NFileName=paste(prefix,".grm.N.bin",sep="")
IDFileName=paste(prefix,".grm.id",sep="")
id = read.table(IDFileName) # read the ID of the gmatrix
n=dim(id)[1]
BinFile=file(BinFileName,"rb")
grm=readBin(BinFile,n=n*(n+1)/2,what=numeric(0),size=size) # generate the fack gmatrix
NFile=file(NFileName,"rb");
if(AllN==T){
N=readBin(NFile,n=n*(n+1)/2,what=numeric(0),size=size)
}else{
N=readBin(NFile,n=1,what=numeric(0),size=size)
}
i=sapply(1:n,sum_i)
return(list(diag=grm[i],off=grm[i],id=id,N=N))
}计算近交系数
--ibc: 会用三种方法计算近交系数.
示例:
gcta64 --bfile test --autosome --make-grm --out grm
这里:
--bfile读取的是plink的二进制文件--autosome是利用常染色体上的所有SNP信息, 这个不能省略--make-grm生成grm矩阵--out生成前缀名
会生成如下三个文件夹:
(base) [dengfei@localhost plink_file]$ ls grm* grm.grm.bin grm.grm.id grm.grm.N.bin
7. 利用构建好的G矩阵, 计算PCA分析
--grm test: 这里的xx是前缀, 它其实包括三个文件:
test.grm.bin, test.grm.N.bin test.grm.id
命令:
gcta64 --grm grm --pca 3 --out out_pca
--grmgrm文件--pcaPCA的数目为3--out结果输出文件
结果生成两个文件:
(base) [dengfei@localhost plink_file]$ ls out_pca.eigenv* out_pca.eigenval out_pca.eigenvec
8. 利用PCA结果画图
在R语言中, 设置好工作路径, 键入如下命令:
dd=read.table("out_pca.eigenvec",header=F)
head(dd)
names(dd) = c("Fid","ID","PC1","PC2","PC3")
plot(dd$PC1,dd$PC2,pch=c(rep(1,112),rep(2,103)),col=c(rep("blue",112),rep("red",103)),main="PCA",xlab="pc1",ylab="pc2")
legend("bottomright",c("TEXT1","TEXT2"),pch=c(rep(1),rep(2)),col=c(rep("blue"),rep("red")))结果:
后记1, 使用示例数据b.ped和b.map使用gcta64做PCA分析
将ped文件, 转化为bed文件
plink --file b --make-bed --out test
生成test.bed, test.bim,test.fam三个文件
构建G矩阵
grm
gcta64 --bfile test --autosome --make-grm --out grm
生成三个文件:
grm.grm.bin grm.grm.id grm.grm.N.bin
生成PCA, 数目为3
gcta64 --grm grm --pca 3
生成两个文件:
gcta.eigenval gcta.eigenvec
作图
dd=read.table("gcta.eigenvec",header=F)
head(dd)
names(dd) = c("Fid","ID","PC1","PC2","PC3")
plot(dd$PC1,dd$PC2,pch=c(rep(1,112),rep(2,103)),col=c(rep("blue",112),rep("red",103)),main="PCA",xlab="pc1",ylab="pc2")
legend("bottomright",c("TEXT1","TEXT2"),pch=c(rep(1),rep(2)),col=c(rep("blue"),rep("red")))结果:
后记2, 使用示例数据b.ped和b.map使用plink做PCA分析
看完gcta, 发现plink也可以构建G矩阵, 也可以进行PCA分析, 本数据使用plink的解决方案:
只用一行代码, 就可以生成PCA的数据, 比gcta64简单太多了.
plink --file b --pca 3
比较两个数据的结果, 可以看出, plink和gcta64结果一致.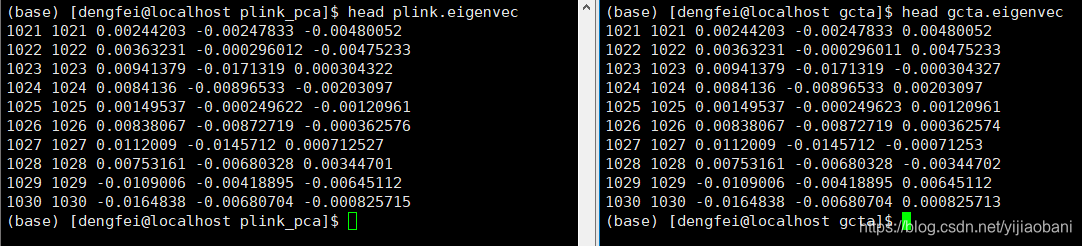
对PCA作图:
结果一致, 因为plink调用的是gcta64的算法, 构建G矩阵, 构建PCA.
福利1
计算gcta64或者plink可以构建矩阵, asreml也支持下三角的G矩阵或者G逆矩阵, 问题来了, 两者怎么联系到一起呢?
这样asreml就可以愉快的进行GBLUP的分析了.
福利2
之前的博客中有提到利用H矩阵构建PCA分析, 那么如何操作呢?
欲听后事如何, 请听下回分解.
公众号后台回复:plink, 获得测试数据:b.ped和b.map, 用于本次分析.
如果您对于数据分析,对于软件操作,对于数据整理,对于结果理解,有任何问题,欢迎联系我。

https://wap.sciencenet.cn/blog-2577109-1175834.html
上一篇:Admixture使用说明文档cookbook
下一篇:blupf90根据G矩阵和H矩阵构建PCA分析以及与Plink以及GCTA的结果对比