博文
Materials Studio官方教程:Forcite——寻找分子在表面上的低能构型【1】
||

目的:演示如何使用分子动力学和几何优化来寻找低能量的极小值。
所用模块:Materials Visualizer、Forcite Plus、QSAR、COMPASS
前提条件:绘制简单分子结构可视化工具使用(Sketching simple molecules Visualizer)教程、有无对称约束下的尿素晶体的几何优化(Geometry optimization of urea with and without symmetry constraints)教程
背景
小分子的势能面可能非常复杂,有许多局部能量极小值和一个全局能量最小值。有几种方法可用于确定全局最小值,包括蒙特卡罗算法和不同形式的分子动力学。淬火分子动力学通过附加的几何优化步骤执行标准分子动力学计算,在该步骤中,对轨迹文件中的每一帧执行几何优化。分子动力学可以对许多不同的低能构型进行有效采样。
介绍
本教程使用Forcite Plus对由小有机分子邻氯苯酚和金属氧化物二氧化钛表面组成的体系执行分子动力学。本教程由金红石相的TiO2晶体,切割二氧化钛表面,再将分子放置在表面上,对其进行优化,并运行淬火分子动力学。可以通过数据表寻找能量最低的构象,最后计算结合能。
本教程包括如下部分:
开始
氯苯酚结构的绘制与优化
构建并优化TiO2表面结构
表面上的分子的平衡位置计算
使用淬火动力学对构型进行采样
计算结合能
注意:为了和本教程中的参数保持一致,可以使用Settings Organizer对话框将工程中所有参数都设置为BIOVIA的默认值。
1、开始
首先启动Materials Studio并创建一个新工程。
打开New Project对话框,输入Cl_phenol作为工程名,单击OK按钮。
新工程将以Cl_phenol为工程名显示于Project Explorer中。
2、氯苯酚结构的绘制与优化
绘制邻氯苯酚分子结构。
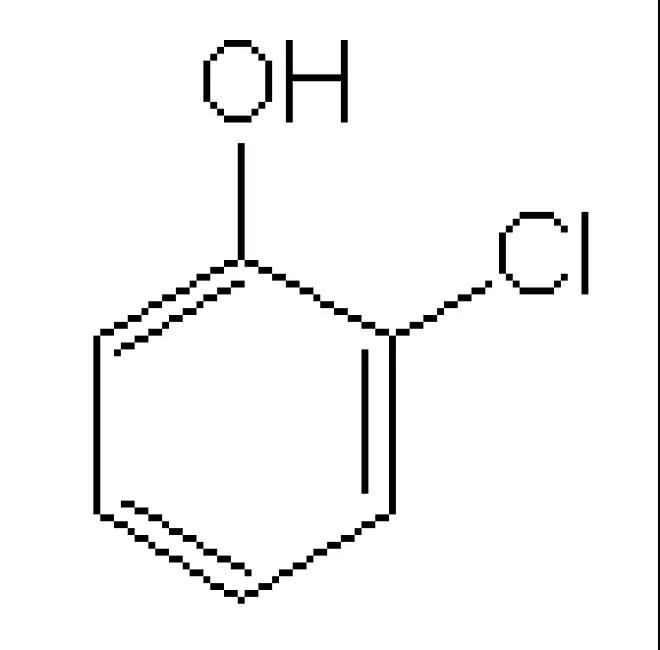
创建一个新的3D原子文档3D Atomistic Document。绘制一个邻氯苯酚分子,随后使用Adjust Hydrogen和Clean工具。
在Project Explorer,将3D原子文档重命名为chlorophenol.xsd。
所绘制的分子的结构并不精确,因此需要使用经典模拟工具进行几何优化。经典模拟工具需要一个力场,用不同类型原子之间的距离、角度等函数的总和来描述原子之间的力。这里将使用Forcite经典模拟引擎和COMPASS力场。
单击Modules工具条上的Forcite按钮 ,在下拉列表中选择Calculation,或在菜单栏中选择Modules | Forcite | Calculation。
,在下拉列表中选择Calculation,或在菜单栏中选择Modules | Forcite | Calculation。
将打开Forcite Calculation对话框。
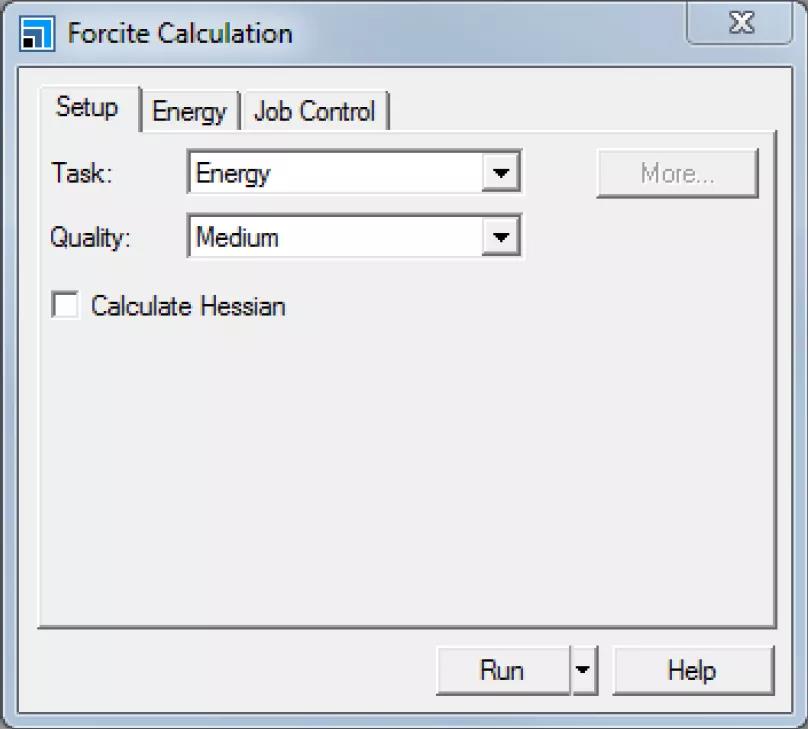
Forcite Calculation对话框的Setup选项卡
在该对话框中,可以选择执行何种计算任务,计算精度Quality根据所选择的计算任务控制不同的参数。在Energy选项卡中,可以选择力场、电荷计算方法,并自定义对非键项的处理方式。
将Task修改为Geometry Optimization。在Energy选项卡中,将Forcefield修改为COMPASSIII。
COMPASS是一个已经计算了电荷的、经过充分验证的力场。因此,当选择COMPASS时,Charges选项将自动更改为由力场分配Forcefield assigned。非键设置默认为基于原子截断Atom based。这适用于小分子,但当稍后引入TiO2表面时,会将其更改为Ewald。
单击Run按钮。
这将启动计算任务并将其显示在Job Explorer中。计算任务将在Job Explorer中创建一个名为chlorophenol Forcite GeomOpt的新文件夹。计算任务完成后,将显示提示,计算结果文件夹中包含6个文件。
chlorophenol.xsd:包含初始结构优化后的几何结构
chlorophenol - Calculation:包含计算任务设置的xml状态文件。单击此文件将打开Forcite Calculation对话框,其中包含计算中指定的设置。
chlorophenol Convergence.xcd:能量和梯度法线的变化图。
chlorophenol Energies.xcd:焓变图。急剧下降表明初始绘制的较粗糙的几何构型已成功优化。
Status.txt:包含实时更新状态。
chlorophenol.txt:初始设置的文本版本,以及初始和最终结构的能量项分解。
3、构建并优化TiO2表面结构
Materials Studio的结构库中包含大量各种结构,其中包含许多常见的金属氧化物。存在三种晶型的二氧化钛。在这里,使用金红石相。
在Project Explorer中,右键单击工程根目录,从弹出的快捷菜单中选择Import...,打开Import Document对话框。导航至Structures\metal-oxides并导入TiO2_rutile.xsd文件。
在切割表面之前,优化晶体结构的几何构型。
在Forcite Calculation对话框的Setup选项卡中,单击More...按钮,打开Forcite Geometry Optimization对话框。选择Optimize cell复选框,关闭对话框。
作为一个三维周期性结构,可以选择优化晶胞和原子位置,或仅优化原子位置。在本例中,同时优化晶胞和原子位置。
在Properties Explorer中,将Filter改为Lattice 3D。
二氧化钛的金红石相属于P42/MNM空间群。Forcite可以在带有对称性约束的条件下优化结构。
单击Run按钮,关闭Forcite Calculation对话框。
将创建一个新文件夹,TiO2_rutile Forcite GeomOpt。当计算结束时,新文件夹包含与氯苯酚文件夹中类似的一系列文件。另外,由于这是一个周期性结构,Forcite也给出了晶胞参数和密度的变化。
注意:TiO2_rutile.txt输出文件会报告某些能源贡献缺少参数。这些不会影响本教程的结果,但可以通过手动分配力场类型,然后删除结构中的所有化学键来避免这些提示消息。聚合物与金属氧化物表面相互作用(Polymer interactions with a metal oxide surface)教程的第2步使用这种方法。
现在已经优化了结构,切割晶体并暴露活性110表面。
提示:如果不知道活性表面,可以使用Morphology模块进行搜索。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
打开优化过的TiO2_rutile.xsd文件。选择Build | Surfaces | Cleave Surface,打开Cleave Surface对话框。将Cleave plane更改为1 1 0,按下TAB键。
在TiO2_rutile.xsd中,将显示一个蓝色盒子,以蓝色实线显示切割平面,点线表示切割深度。
使用微调按钮将Fractional Top增加到0.4,Fractional Thickness增加到3.0。单击Cleave按钮,并关闭对话框。
将打开一个新的3D原子文档TiO2_rutile (1 1 0).xsd。表面由白色矩形表示,包含钛原子和氧原子。
现在要优化表面。由于已经优化了块体晶体结构,因此可以固定底层的几何构型,因为它们表示块体结构。
单击3D Viewer Selection Mode工具。重新定位结构的方向,使得表面在屏幕顶端。选择底部的Ti和O原子混合层以及其正上方和下方的O原子层部分。从菜单栏中选择Modify | Constraints。
打开Edit Constraints对话框。
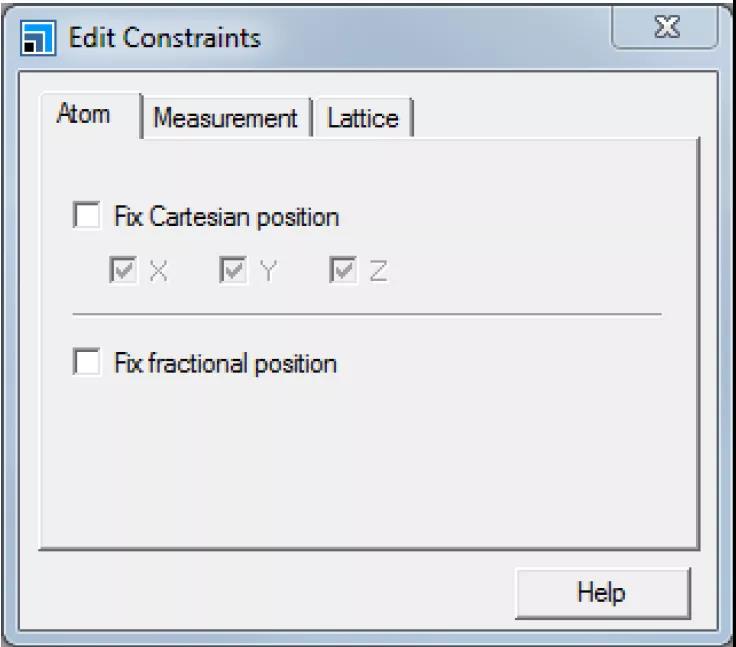
Edit Constraints对话框的Atom选项卡
勾选Fix Cartesian position复选框,关闭对话框。
单击3D Viewer以取消选中所选部分。右键单击3D Viewer,从弹出的快捷菜单中选择Display Style,打开Display Style对话框。在Atom选项卡的Coloring部分,将Color by选项更改为Constraint。
结构应与下图类似,被固定的原子显示为红色,未被固定的原子显示为灰色。
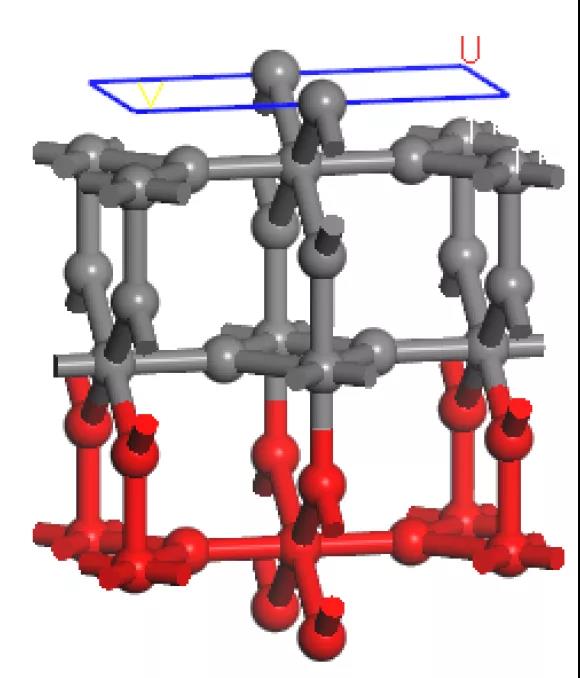
以球棍模型显示的TiO2表面结构
在Display Style对话框,将Color by选项重新改回Element,关闭对话框。
现在将优化表面结构。
打开Forcite Calculation对话框,单击Run按钮。
将再次显示包含结果文件的新文件夹。本部分的最后一步是确定表面的大小。表面必须足够大,以容纳有机分子,同时尽量减少与镜像分子之间的相互作用。因此,首先使用距离监视工具计算氯酚分子的长度尺寸。
打开优化过的chlorophenol.xsd文件。单击Measure/Change按钮
![]() 的下拉箭头,选择Distance。选择Cl原子及与之对位的H原子。
的下拉箭头,选择Distance。选择Cl原子及与之对位的H原子。
将显示一个值约为5.6 Å的距离测量。假设每个分子周围有2 Å区域,则分子间相互作用可以达到最小化,这表明单位晶胞大小为5.6 + 2 × 2或约为9.6 Å。现在,需要确定必须生成多少个当前表面的副本才能实现此目的。
使得优化好的金红石相表面为当前文档。在Properties Explorer中,将Filter更改为Lattice 2D。
U和V的晶胞参数分别约为3.0和6.3 Å。因此,3 × 2的超晶胞对于容纳有机分子是足够的。
从菜单栏中选择Build | Symmetry | Supercell。将U更改为3,V更改为2。单击Create Supercell按钮,关闭对话框。
已经建立了表面结构。在继续下一步之前,保存工程。
从菜单栏中选择File | Save Project。
Materials Studio是久负盛名计算模拟软件,问世20余年来,经过不断地迭代优化,使其功能异常强大,极易上手,初学者只需通过简单的参数设置和点击鼠标就能完成DFT计算。其计算可靠性久经考验,备受Nature、Science等顶级期刊认可。
华算科技和Materials Studio官方代理深圳浦华系统联合推出Materials Studio建模、计算、分析课程。课程专为零基础学员设计,沿着理论讲解、模型搭建、性质计算、结果分析层层递进讲解,带你快速入门DFT计算。课程极度注重实操,全程线上直播,提供无限回放,课程群在线答疑。(详情点击下方图片跳转)
识别下方二维码报名,或者联系手机13005427160。

https://wap.sciencenet.cn/blog-2531381-1319299.html
上一篇:Discovery Studio 的扩展性
下一篇:Materials Studio官方教程:Forcite——寻找分子在表面上的低能构型【2】
全部作者的其他最新博文
- • Materials Studio官方教程:Mesocite——磷脂双分子层的粗颗粒化分子动力学【1】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【3】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【2】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【1】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【3】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【2】