博文
Materials Studio官方教程:Forcite——有无对称约束下的尿素晶体的几何优化
||

目的:说明了使用Forcite模块优化几何结构时施加对称性的影响。
所用模块:Materials Visualizer、Forcite、COMPASS
背景
Materials Studio软件中的Forcite模块是一款经典的分子力学工具,由BIOVIA公司的科学家和软件工程师设计,用于执行一系列计算任务,包括对单分子和周期性体系的快速能量计算和几何优化。在晶体结构的几何优化中,Forcite可保留体系的对称性,这在与确定结构相关的计算中尤为重要。
介绍
在确定晶体结构时,对称要素的正确性以及结构处于能量最小值这两点均十分重要。这意味着在没有对称约束的情况下,优化的结构在随后的几何优化过程中不应发生太大变化。
本教程旨在演示在执行晶体结构的几何优化时施加对称性的作用。在本教程中,使用尿素的晶体结构,尿素是一种广泛使用的化学和医药中间体。
本教程包括如下部分:
开始
对施加对称的结构和单位晶胞进行几何优化
删除对称性并执行进一步的几何优化
比较两次运行的结果
注意:为了和本教程中的参数保持一致,可以使用Settings Organizer对话框将工程中所有参数都设置为BIOVIA的默认值。
1、开始
首先启动Materials Studio并创建一个新工程。
打开New Project对话框,输入urea作为工程名,单击OK按钮。
新工程将以urea为工程名显示于Project Explorer中。导入体系的晶体结构来研究尿素。
单击Import按钮![]() ,打开Import Document对话框。导航至Structures/molecular-crystals/misc文件夹,双击urea.xsd文件。
,打开Import Document对话框。导航至Structures/molecular-crystals/misc文件夹,双击urea.xsd文件。
尿素的晶体结构即显示于一个名为urea.xsd的3D原子文档中。验证结构的对称性。
单击Symmetry工具条上的Show Symmetry按钮![]() ,或菜单栏中的Build | Symmetry | Show Symmetry。
,或菜单栏中的Build | Symmetry | Show Symmetry。
打开Symmetry对话框。
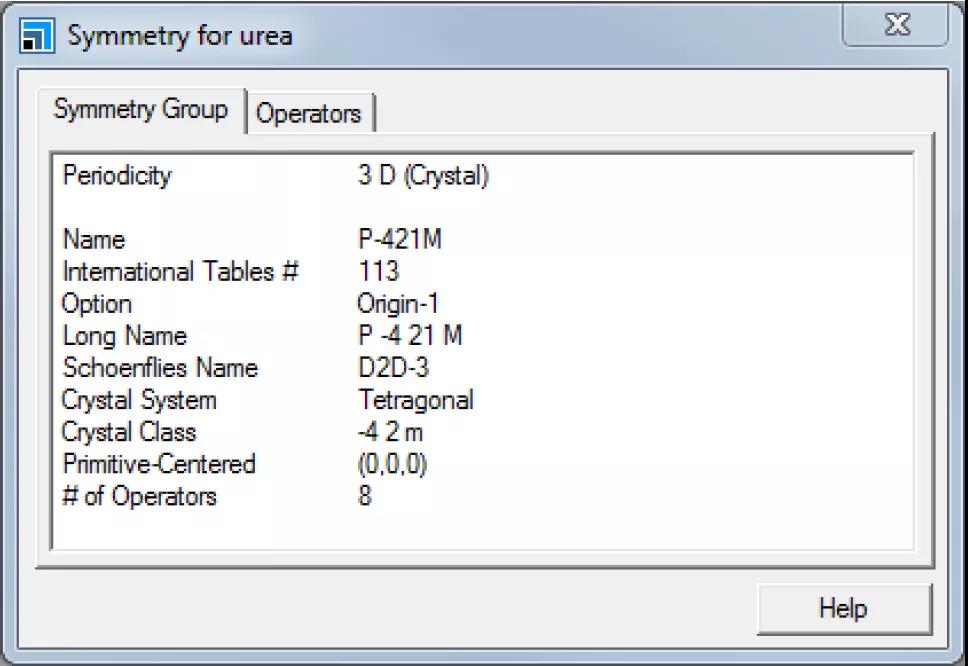
Symmetry对话框
即显示了晶体的空间群为P-421M。
2、对施加对称的结构和单位晶胞进行几何优化
下一步是使用COMPASS力场对尿素进行几何优化。
单击Modules工具条上的Forcite按钮![]() ,在下拉列表中选择Calculation,或在菜单栏中选择Modules | Forcite | Calculation。
,在下拉列表中选择Calculation,或在菜单栏中选择Modules | Forcite | Calculation。
将打开Forcite Calculation对话框。
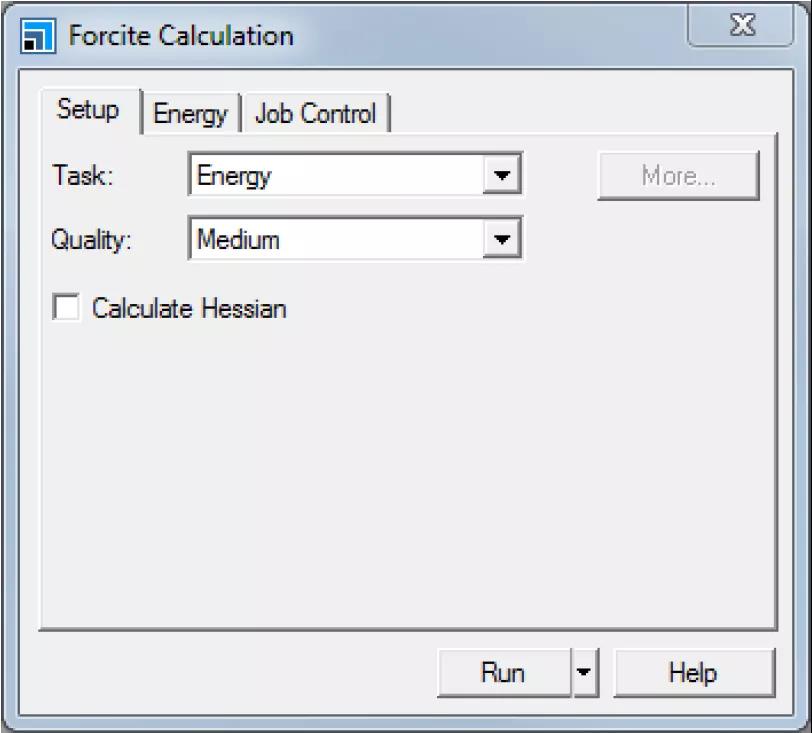
Forcite Calculation对话框的Setup选项卡
将Task由Energy修改为Geometry Optimization,将Quality设置为Fine。
单击More...按钮,打开Forcite Geometry Optimization对话框。勾选Optimize cell复选框,关闭对话框。
在Energy选项卡中,从Forcefield下拉列表里选择COMPASSIII。保持所有其他设置不变。
单击Run按钮并关闭对话框。
将在Project Explorer中打开一个名为urea Forcite GeomOpt的新文件夹。由于结构库中的结构已接近最优,因此计算只需不到1分钟即可完成,即达到规定的收敛标准。计算完成后,新文件夹接近顶部的urea.xsd文档包含优化的结构。
urea Forcite GeomOpt文件夹中还有其他六个文档。urea.txt文档包含计算任务的所有文本信息。包括初始和最终结构的结构和能量参数值。Status.txt文档包含最终计算任务的执行状态。能量图urea Energy显示了优化过程中总能量的变化。收敛图urea Convergence显示了收敛标准的变化,即能量变化、梯度范数和应力范数与优化步骤的函数关系。一旦满足所有设定的条件,模拟就会停止。单位晶胞Cell图显示了晶胞参数的变化,以及密度Density图显示了体系密度的变化。
打开图表文档urea Energies.xcd。
该文件应与下图相似。
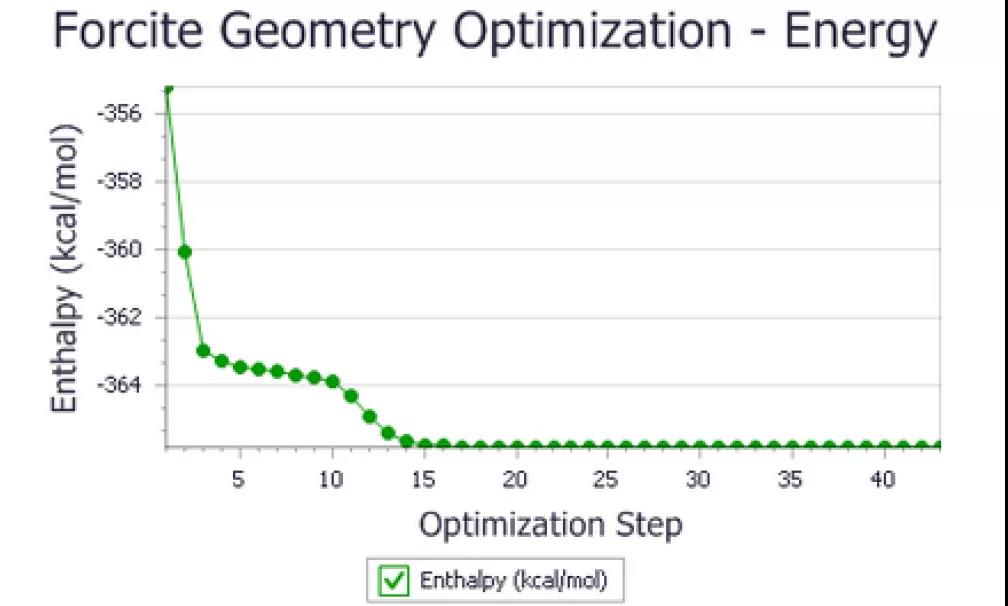
确保对称性未发生变化。
使得urea Forcite GeomOpt/urea.xsd为当前文档,打开对称性Symmetry对话框。
空间群仍为P-421M。在完成教程该部分之前,保存工程,并清理工作区。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
3、删除对称性并执行进一步的几何优化
现在在无对称性限制的情况下,重复上述步骤。首先导入尿素结构的另一个副本文件。
打开Import Document对话框,导航至Structures/molecular-crystals/misc/,导入urea.xsd。
将打开另一个包含尿素结构的3D原子文档,名为urea (2).xsd。去除晶体结构的对称性。
从菜单栏中选择Build | Symmetry | Make P1。
确定现在的对称性为P1。
在Symmetry对话框中,确定空间群Space group为P1。关闭对话框。
为反映对称性的变化,将urea (2).xsd重命名。
在Project Explorer右键单击urea (2).xsd,从弹出的下拉列表中选择Rename。将名字改成urea_P1。
下一步是对urea_P1进行几何优化,使用与之前相同的设置。
打开Forcite Calculation对话框。
Forcite保留所有之前使用的参数。由于将以与之前相同的参数设置运行计算,因此无需进行任何更改。
单击Run按钮,关闭对话框。
将在Project Explorer中打开一个新的名为urea_P1 Forcite GeomOpt的对话框。计算仍然完成的非常迅速。当计算任务结束后,再次保存工程,并关闭所有打开的窗口。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
4、比较两次运行的结果
最后一步是比较两次计算的结果。
找到并打开输出文本文档urea.txt和urea_P1.txt,并将其打开。向下滚动到最终结构的总能量Total energy。
得到的值几乎相同,为-365.83 kcal/mol。现在比较两种结构的晶胞优化结果。
找到并打开图表文档urea Cell.xcd和urea_P1 Cell.xcd。
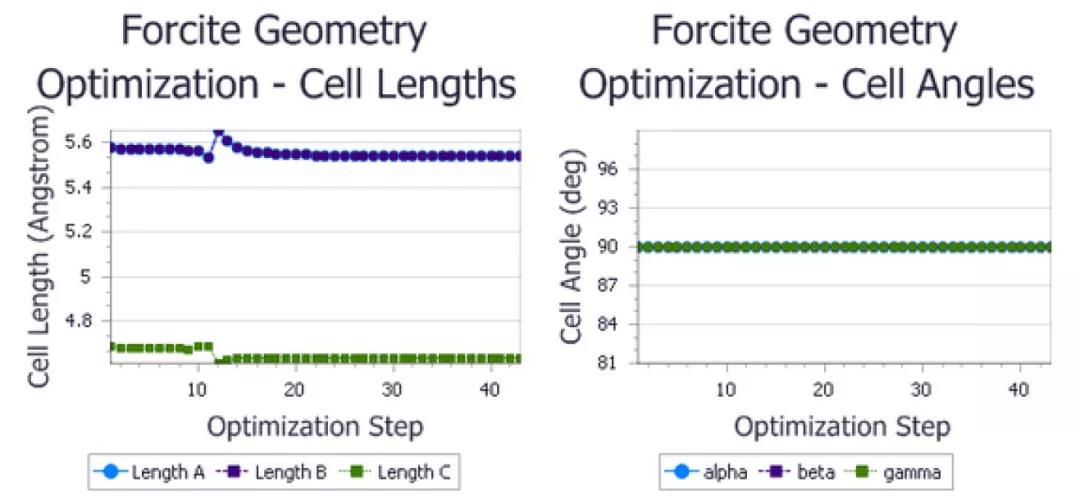
P-421M对称性下优化得到的尿素晶胞参数
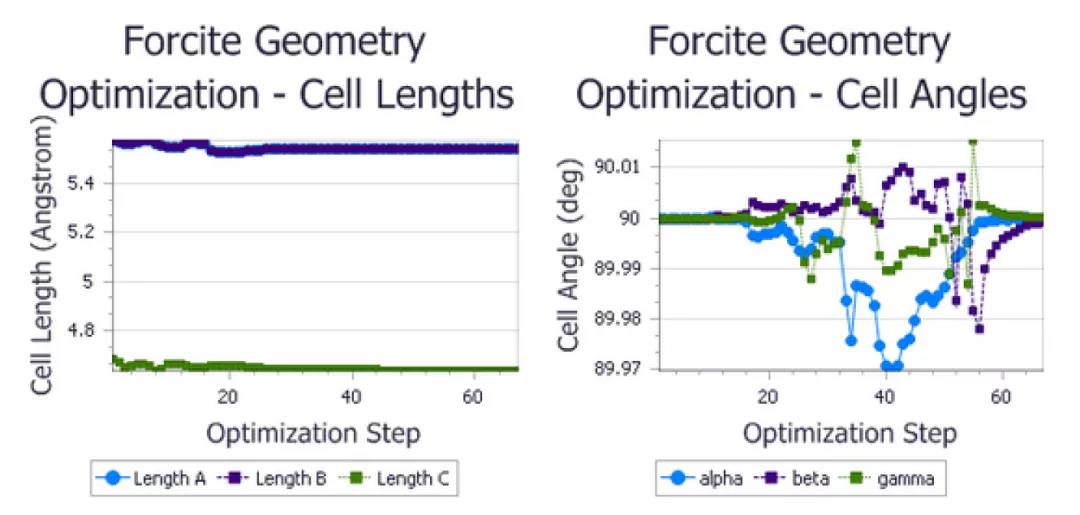
P1对称性下优化得到的尿素晶胞参数
在保持尿素晶体原有对称性的情况下,晶格长度略有减少,并且由于对称约束,晶格夹角保持在90°不变。对于对称性为P1结构,在没有对称性约束的情况下,晶格夹角在模拟过程中会发生变化。虽然精确变化对计算细节非常敏感,但角度最终再次收敛到90°。所以,总的来说,这两种情况下的晶格夹角也非常相似。
找到并打开图表Density.xcd和urea_P1 Density.xcd。
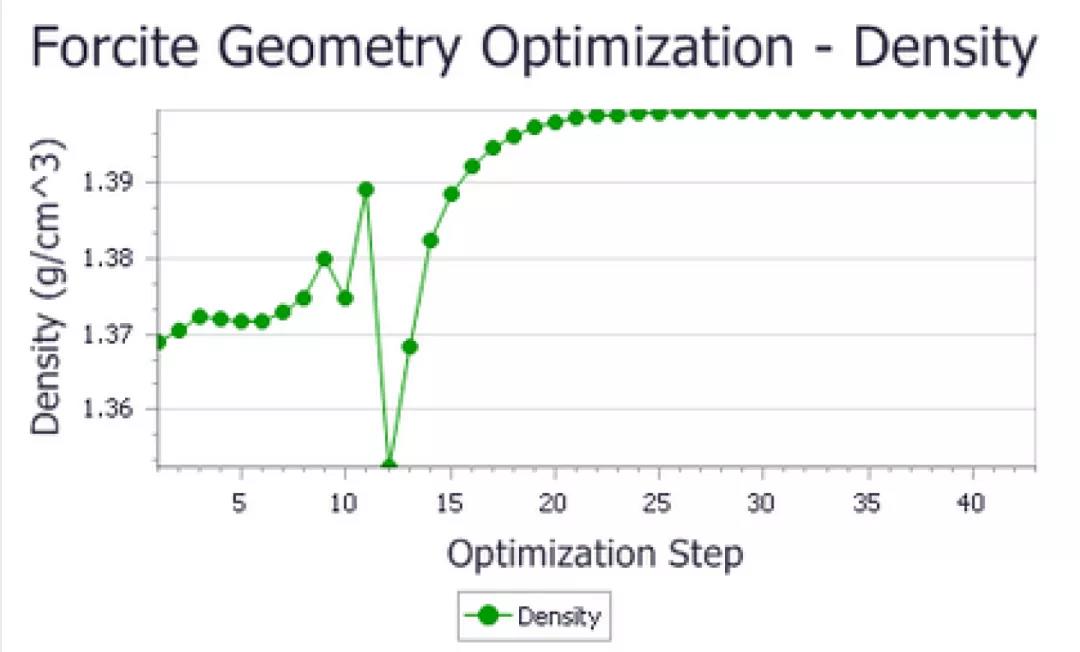
P-421M对称性下优化得到的尿素密度
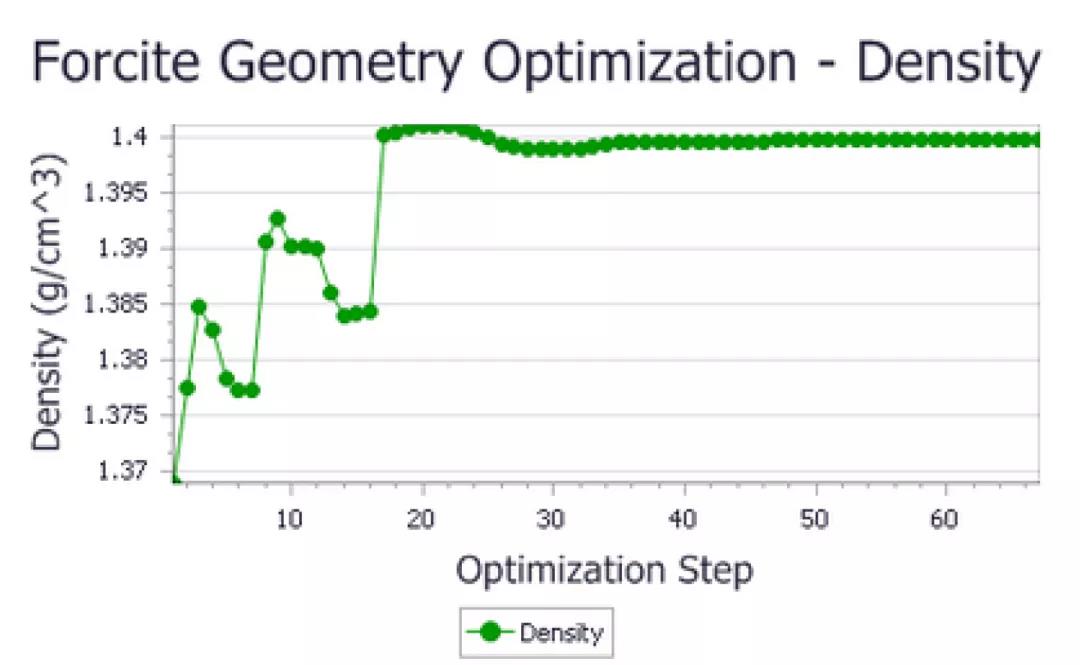
P1对称性下优化得到的尿素密度
在这两种情况下,由于优化,尿素单位晶胞的密度略有增加。同样,精确变化取决于计算细节,但最终值略低于1.40 g/cm3。
可以得出这样的结论:在是否施加对称性的情况下,几何优化的过程虽然有较大差异,但最终两种情况下的计算结果都会收敛到非常相似的值。
因此,在Forcite几何优化过程中,对称要素会保持不变,这些对称要素的存在与否会严重影响计算过程。但是如本例中施加或未施加对称约束的情况下的计算结果,一旦一个结构接近它的最小能量构象,它就可以收敛,并得出非常相似的晶胞参数和能量值。
本教程到此结束。
本教程对应视频将在杨站长视频号、华算科技B站同步推送,敬请各位关注。
Materials Studio是久负盛名计算模拟软件,问世20余年来,经过不断地迭代优化,使其功能异常强大,极易上手,初学者只需通过简单的参数设置和点击鼠标就能完成DFT计算。其计算可靠性久经考验,备受Nature、Science等顶级期刊认可。
华算科技和Materials Studio官方代理深圳浦华系统联合推出Materials Studio建模、计算、分析课程。课程专为零基础学员设计,沿着理论讲解、模型搭建、性质计算、结果分析层层递进讲解,带你快速入门DFT计算。课程极度注重实操,全程线上直播,提供无限回放,课程群在线答疑。(详情点击下方图片跳转)
识别下方二维码报名,或者联系手机13005427160。

https://wap.sciencenet.cn/blog-2531381-1318514.html
上一篇:Discovery Studio | 基于小分子的药物发现和设计模块
下一篇:Materials Studio官方教程:Forcite——氢在钨表面的物理吸附
全部作者的其他最新博文
- • Materials Studio官方教程:Mesocite——磷脂双分子层的粗颗粒化分子动力学【1】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【3】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【2】
- • Materials Studio官方教程:Kinetix——CO在Pt(1 1 1)表面氧化的多位点模拟【1】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【3】
- • Materials Studio官方教程:Kinetix——蒙特卡洛方法模拟CO的氧化【2】