博文
新型非成瘾化合物无需镇静即可缓解疼痛
 精选
精选
||
新型非成瘾化合物无需镇静即可缓解疼痛
诸平
据美国加州大学旧金山分校(University of California, San Francisco简称UCSF)2022年11月8日报道,新型非成瘾化合物无需镇静即可缓解疼痛(New Non-Addictive Compound Alleviates Pain Without Sedation)。研究人员认为这种新分子不太可能会让人上瘾。
据加州大学旧金山分校(UCSF)的研究人员称,这种新分子是最有前景的麻醉剂替代品。UCSF研究人员最近领导的一项研究发现,一组新发现的分子可以减轻小鼠的疼痛,而不具有限制使用的阿片类药物(opiates)的镇静作用。这些分子作用于与可乐定(clonidine)和右美托咪啶(dexmedetomidine)相同的受体上,这两种镇静剂在医院中经常使用,但它们在化学上没有关系,可能不会上瘾。相关研究结果于2022年9月30日已经在《科学》(Science)杂志网站发表——Elissa A. Fink, Jun Xu, Harald Hübner, Joao M. Braz, Philipp Seemann, Charlotte Avet, Veronica Craik, Dorothee Weikert, Maximilian F. Schmidt, Chase M. Webb, Nataliya A. Tolmachova, Yurii S. Moroz, Xi-Ping Huang, Chakrapani Kalyanaraman, Stefan Gahbauer, Geng Chen, Zheng Liu, Matthew P. Jacobson, John J. Irwin, Michel Bouvier, Yang Du, Brian K. Shoichet, Allan I. Basbaum, Peter Gmeiner. Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor. Science, 30 September 2022. Vol 377, Issue 6614. DOI: 10.1126/science.abn7065. https://www.science.org/doi/10.1126/science.abn7065
参与此项研究的除了来自美国UCSF的研究人员之外,还有美国斯坦福大学医学院(Stanford University School of Medicine, CA, USA)、美国北卡罗来纳大学教堂山医学院(University of North Carolina at Chapel Hill School of Medicine, NC, USA);中国深圳香港中文大学(Chinese University of Hong Kong, Shenzhen, China)、德国埃尔朗根-纽伦堡大学全称为弗里德里希-亚历山大 埃尔朗根-纽伦堡大学(Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany)、加拿大蒙特利尔大学(Université de Montréal, QC, Canada.)、乌克兰国家科学院(National Ukrainian Academy of Science, Kyiv, Ukraine)以及乌克兰基辅国立塔拉斯·舍甫琴科大学(National Taras Shevchenko University of Kyiv, Ukraine)的研究人员。
该研究的四位通讯作者之一、药学院教授布莱恩·肖切特(Brian Shoichet)博士说:“我们发现,分离与这种受体相关的镇痛和镇静作用是可能的。这使它成为药物开发的一个非常有希望的目标。”
这项研究是美国国防部高级研究计划局(Defense Advanced Research Projects Agency简称DARPA)一项为期5年拨款的一部分,开始于COVID-19爆发前不久,目的是发现可以与阿片类药物(opioids)一起使用或联合使用的有效止痛药。
该研究汇集了来自不同领域的学者:布莱恩·肖切特的合著者包括UCSF解剖学主席艾伦·巴斯鲍姆(Allan Basbaum)博士、来自德国埃尔朗根-纽伦堡大学的化学家彼德·格迈纳(Peter Gmeiner)博士、香港中文大学结构生物学家杜阳(Yang Du音译)博士以及加拿大蒙特利尔大学分子生物学家米歇尔·布维尔(Michel Bouvier)博士。
艾伦·巴斯鲍姆说:“我们一起从最基本的层面着手,鉴定出可能相关的新分子,然后证明它们确实是相关的。这种情况并不常见。”
从3亿分子遴选出6种(6 Molecules Out of 300 Million)
艾伦·巴斯鲍姆在实验室中研究了这种称为α2A的肾上腺素能受体(adrenergic receptor),发现它与缓解疼痛有关,他鼓励布莱恩·肖切特寻找能激活α2A的物质。
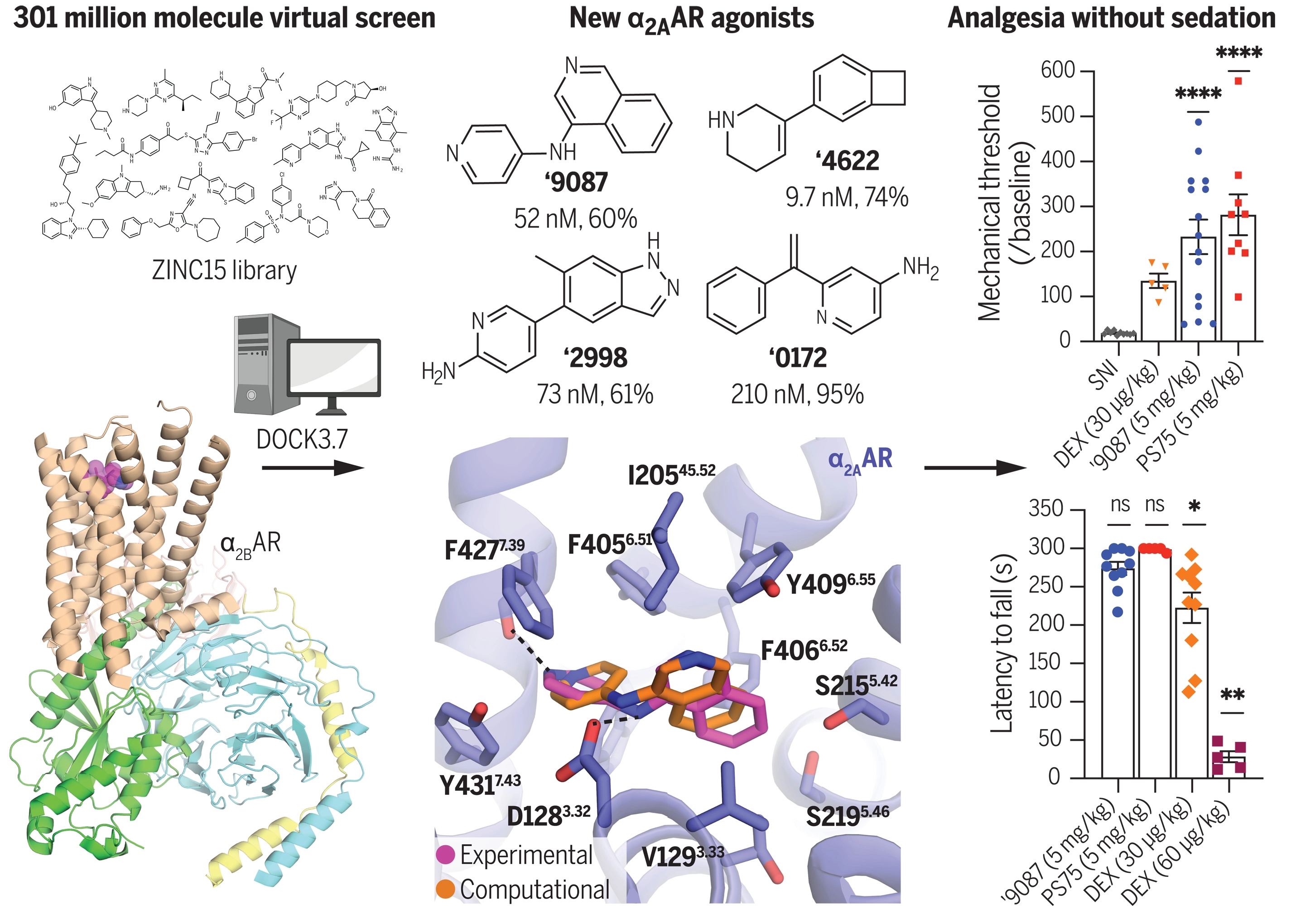
布莱恩·肖切特通过计算扫描了一个包含3亿多种分子的虚拟库(virtual library),开始寻找那些能与受体牢固结合的分子,排除那些对小受体来说太大的分子。剩下的数万种分子实际上一个接一个地“对接”到受体的计算机模型上。
经过一系列的测试,布莱恩·肖切特将范围从最初的48种分子减少到了6种分子,这取决于它们是如何与培养的人类和小鼠细胞中的受体结合的。最后的6种分子中的每一种都在3种不同的小鼠模型上进行了急性和慢性疼痛的测试,并且在所有3种情况下都成功地减轻了疼痛。
这些止痛分子(pain-relieving molecules)来自不同的化学家族,它们也是全新的。它们中的任何一种分子之前都尚未合成过。
布莱恩·肖切特说,虽然老的药物,如右美托咪啶(dexmedetomidine),可以激活广泛的神经通路(neuronal pathways),但新分子只会选择性地激活其中的一部分。这些分子也集中在大脑中,并与受体紧密结合,使它们成为进一步发育的良好候选者。
为1/5的美国人带来希望(Hope for 1 in 5 Americans)
艾伦·巴斯鲍姆警告说,这些化合物在进行临床试验之前可能还需要几年的研究。研究人员还不清楚新分子可能产生的副作用,以及长期使用是否会产生意想不到的后果。然而,他认为这种化合物不太可能上瘾。他说:“药物滥用是在药物产生回报时发生的,但我们没有发现任何证据。”
虽然阿片类药物明显有助于缓解手术或癌症带来的疼痛,但艾伦·巴斯鲍姆指出,在5000万患有慢性疼痛的美国人中,大多数患有其他疾病,如背部受伤、关节疼痛和炎症疾病,这些往往是药物无法缓解的。新的止痛剂可能完全改变这些患者的前景,为其带来福音。
他说,“如果我们能创造出一种药物,与低剂量的阿片类药物结合使用,那将是我们的梦想。这种需求是巨大的。”
该研究由美国国防高级研究计划局(Defense Advanced Research Projects Agency: HR0011-19-2-0020)、美国国立卫生研究院(National Institutes of Health: R35GM122481; R01GM133836; US R35 NS097306)、美国国家心理卫生研究所(National Institute of Mental Health: HHSN-271-2018-00023-C)、中国深圳市科学技术和创新委员会(Science, Technology and Innovation Commission of Shenzhen Municipality: JCYJ20200109150019113)、深圳重点实验室项目(Shenzhen Key Lab Project: ZDSYS20190902093417963)、德国科学基金会(Deutsche Forschungsgemeinschaft: GRK 1910)以及加拿大卫生研究院(Canadian Institutes of Health Research: FN-148431)的资助。
上述介绍,仅供参考。欲了解更多信息,敬请注意浏览原文或者相关报道。
The serious problems associated with opioid addiction have motivated the search for non-opioid pain-relief drugs. The α2A-adrenergic receptor (α2AAR) is a validated pain receptor and is targeted by dexmetadomine, a drug used in hospitals but unsuitable for broader use because it causes sedation and is not orally bioavailable. Fink et al. screened more than 300 million virtual molecules and identified agonists that bind α2AAR with reasonable affinity and are structurally unrelated to known agonists. Experimental structures of two of the compounds bound to α2AAR allowed optimization to improve potency. The optimized compounds were effective in a neuropathic pain model without causing sedation, making them promising leads for further development. —VV
Because nonopioid analgesics are much sought after, we computationally docked more than 301 million virtual molecules against a validated pain target, the α2A-adrenergic receptor (α2AAR), seeking new α2AAR agonists chemotypes that lack the sedation conferred by known α2AAR drugs, such as dexmedetomidine. We identified 17 ligands with potencies as low as 12 nanomolar, many with partial agonism and preferential Gi and Go signaling. Experimental structures of α2AAR complexed with two of these agonists confirmed the docking predictions and templated further optimization. Several compounds, including the initial docking hit ‘9087 [mean effective concentration (EC50) of 52 nanomolar] and two analogs, ‘7075 and PS75 (EC50 4.1 and 4.8 nanomolar), exerted on-target analgesic activity in multiple in vivo pain models without sedation. These newly discovered agonists are interesting as therapeutic leads that lack the liabilities of opioids and the sedation of dexmedetomidine.
https://wap.sciencenet.cn/blog-212210-1363042.html
上一篇:一种由石墨烯制成的新型量子元件
下一篇:研究团队创造出热发电晶体