博文
Seurat的单细胞免疫组库分析来了!
||
小剧场:
我:老板,你听说没有楼上做的单细胞实验加了VDJ分析? 老板:VCD分析?哦,,我早就听过了! 我:老板,是VDJ分析? 老板:VDJ分析,哦对对就是VDJ分析,,,我早就说咱们也应该做了,你看看,又迟了一步,都怪你不提醒我!我瞟了一眼她桌子上的淘宝页面(廉价灯芯裤),默默走开了。。。
其实我在介绍clonotypr (令我惊奇的是,当我把推送推出去的当天,我亲爱的作者就把该包从github撤了下来啊!)时说明过VDJ免疫分析对免疫及抗体产生的重要意义,这也是为什么现在许多做新冠单细胞分析的都会使用5’端测序联用VDJ测序分析。
1. 为什么要进行单细胞免疫组库的分析?
应用方向一:探索肿瘤免疫微环境,辅助免疫治疗。
每个人都拥有一个自己的适应性免疫组库,TCR和BCR通过基因重组和体细胞突变取得多样性,使得我们身体可以识别和抵御各种内部和外部的入侵者。而肿瘤的发生往往躲避了人体T淋巴细胞而产生、增殖和转移。
使用10X Genomics ChromiumTM Single Cell Immune Profiling Solution可以捕捉肿瘤发生时的免疫微环境变化,寻找免疫治疗的靶点,从而辅助免疫治疗更好地抗击肿瘤。
应用方向二:探索自身免疫性疾病和炎症性疾病发生机制,辅助疫苗的研究
自身免疫性疾病发生起始和发展的中心环节被认为是抗原特异性T细胞激活导致的,使用10X Genomics ChromiumTM Single Cell Immune Profiling Solution,可以解析自身免疫性疾病的发病机制,从而为疾病的诊疗提供依据。
应用方向三:移植和免疫重建
器官或者骨髓移植时,经常会诱发宿主的排斥反应,从而发生慢性移植抗宿主病。同种异体反应随机分布在整个T细胞组库的交叉反应,因此延迟T细胞恢复和限制的T细胞受体多样性与异体移植后感染和疾病复发风险增加相关。
而我比较注意的是在疫苗接种前后BCR/TCR CDR3免疫组库的分析,最近medRxiv上发表的有关新冠的文献Immune Cell Profiling of COVID-19 Patients in the recovery stage by Single-cell sequencing中对不同BCR/TCR的VDJ重排进行分析,揭示了针对新冠特异的克隆扩增。
2. 免疫组库主要包括哪几个方面?
T淋巴细胞(T cell)和B淋巴细胞(B cell)主要负责适应性免疫应答,其抗原识别主要依赖于T细胞受体(T cell recptor, TCR)和B细胞受体(B cell recptor, BCR),这两类细胞表面分子的共同特点是其多样性,可以识别多种多样的抗原分子。BCR的轻链和TCRβ链由V、D、J、C四个基因片段组成,BCR的重链和TCRα链由V、J、C三个基因片段组成,这些基因片段在遗传过程中发生重组、重排,组合成不同的形式,保证了受体多样性。其中变化最大的就是CDR3区。
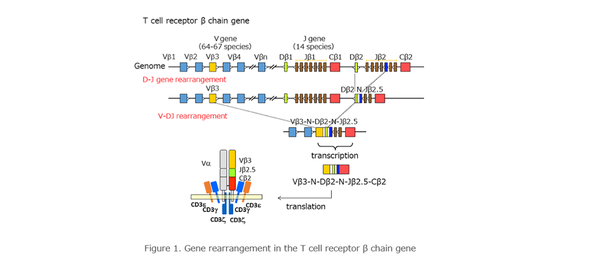
3. 10× Genomics VDJ测序进行cellranger后的输出形式是什么样的?
Outputs: - Run summary HTML: /home/jdoe/runs/sample345/outs/web_summary.html - Run summary CSV: /home/jdoe/runs/sample345/outs/metrics_summary.csv - All-contig FASTA: /home/jdoe/runs/sample345/outs/all_contig.fasta - All-contig FASTA index: /home/jdoe/runs/sample345/outs/all_contig.fasta.fai - All-contig FASTQ: /home/jdoe/runs/sample345/outs/all_contig.fastq - Read-contig alignments: /home/jdoe/runs/sample345/outs/all_contig.bam - Read-contig alignment index: /home/jdoe/runs/sample345/outs/all_contig.bam.bai - All contig annotations (JSON): /home/jdoe/runs/sample345/outs/all_contig_annotations.json - All contig annotations (BED): /home/jdoe/runs/sample345/outs/all_contig_annotations.bed - All contig annotations (CSV): /home/jdoe/runs/sample345/outs/all_contig_annotations.csv - Filtered contig sequences FASTA: /home/jdoe/runs/sample345/outs/filtered_contig.fasta - Filtered contig sequences FASTQ: /home/jdoe/runs/sample345/outs/filtered_contig.fastq - Filtered contigs (CSV): /home/jdoe/runs/sample345/outs/filtered_contig_annotations.csv - Clonotype consensus FASTA: /home/jdoe/runs/sample345/outs/consensus.fasta - Clonotype consensus FASTA index: /home/jdoe/runs/sample345/outs/consensus.fasta.fai - Clonotype consensus FASTQ: /home/jdoe/runs/sample345/outs/consensus.fastq - Concatenated reference sequences: /home/jdoe/runs/sample345/outs/concat_ref.fasta - Concatenated reference index: /home/jdoe/runs/sample345/outs/concat_ref.fasta.fai - Contig-consensus alignments: /home/jdoe/runs/sample345/outs/consensus.bam - Contig-consensus alignment index: /home/jdoe/runs/sample345/outs/consensus.bam.bai - Contig-reference alignments: /home/jdoe/runs/sample345/outs/concat_ref.bam - Contig-reference alignment index: /home/jdoe/runs/sample345/outs/concat_ref.bam.bai - Clonotype consensus annotations (JSON): /home/jdoe/runs/sample345/outs/consensus_annotations.json - Clonotype consensus annotations (CSV): /home/jdoe/runs/sample345/outs/consensus_annotations.csv - Clonotype info: /home/jdoe/runs/sample345/outs/clonotypes.csv - Barcodes that are declared to be targeted cells: /home/jdoe/runs/sample345/out/cell_barcodes.json - Loupe V(D)J Browser file: /home/jdoe/runs/sample345/outs/vloupe.vloupe
首先我们来看看web.html对整个测序质量的评估:
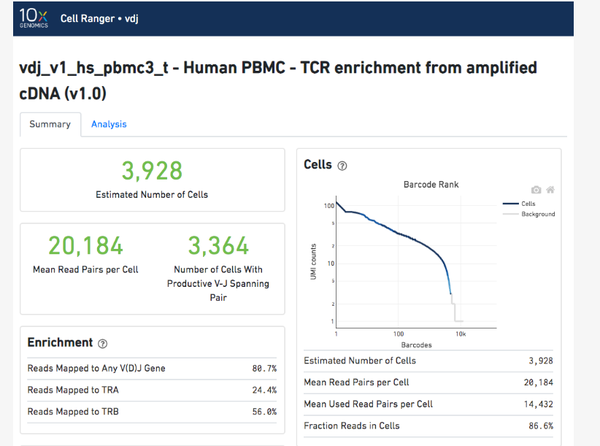
我们看到在Enrichment中reads映射到VDJ基因的比例为80.7%,其中TRA/TRB代表TCR α/β链 ,map到TRA的比例为24.4%,map到TRB的比例为56%。当然,后面也会有蛮多指标的,比如VDJ注释,VDJ质量及表达等。。。
当然我们也会有很多表格,其中最重要的表格为contig_annotation和clonotype:
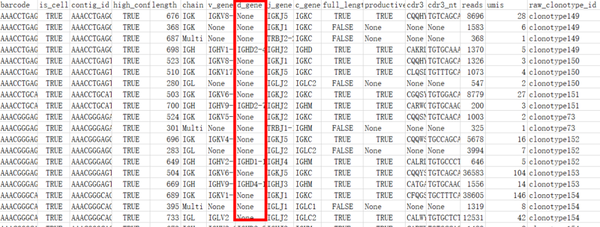
contig_annotation(BCR示例)
上面表格中的IGH和IGK/IGL代表BCR H和BCR L链 。看到这个表格,我第一反应其实是为什么D区基因(d_gene)多数均为None,主要原因还是D区通常较短又突变较多,因技术限制而常常捕捉不到。数据中也提供了CDR3的蛋白序列和核苷酸序列。

clonotype(TCR示例)
从以上数据可以看出,有部分克隆是由单链决定的。
那么如何将VDJ的克隆表型和scRNA-seq结合起来呢?其实大佬已经回答了这个问题:

add_clonotype <- function(tcr_location, seurat_obj){
tcr <- read.csv(paste(tcr_folder,"filtered_contig_annotations.csv", sep=""))
# Remove the -1 at the end of each barcode.
# Subsets so only the first line of each barcode is kept,
# as each entry for given barcode will have same clonotype.
tcr$barcode <- gsub("-1", "", tcr$barcode)
tcr <- tcr[!duplicated(tcr$barcode), ]
# Only keep the barcode and clonotype columns.
# We'll get additional clonotype info from the clonotype table.
tcr <- tcr[,c("barcode", "raw_clonotype_id")]
names(tcr)[names(tcr) == "raw_clonotype_id"] <- "clonotype_id"
# Clonotype-centric info.
clono <- read.csv(paste(tcr_folder,"clonotypes.csv", sep=""))
# Slap the AA sequences onto our original table by clonotype_id.
tcr <- merge(tcr, clono[, c("clonotype_id", "cdr3s_aa")])
# Reorder so barcodes are first column and set them as rownames.
tcr <- tcr[, c(2,1,3)]
rownames(tcr) <- tcr[,1]
tcr[,1] <- NULL
# Add to the Seurat object's metadata.
clono_seurat <- AddMetaData(object=seurat_obj, metadata=tcr)
return(clono_seurat)
}怎么用呢?举个栗子吧:
数据下载
download.file("https://bioshare.bioinformatics.ucdavis.edu/bioshare/download/iimg5mz77whzzqc/vdj_v1_mm_balbc_pbmc.zip", "vdj_v1_mm_balbc_pbmc.zip")#这是小鼠的PBMC数据加载R包
library(Seurat) library(cowplot)
加载数据
## Cellranger
balbc_pbmc <- Read10X_h5("vdj_v1_mm_balbc_pbmc/vdj_v1_mm_balbc_pbmc_5gex_filtered_feature_bc_matrix.h5")
s_balbc_pbmc <- CreateSeuratObject(counts = balbc_pbmc, min.cells = 3, min.features = 200, project = "cellranger")提取线粒体基因
s_balbc_pbmc$percent.mito <- PercentageFeatureSet(s_balbc_pbmc, pattern = "^mt-")
增加T和B细胞的克隆信息
add_clonotype <- function(tcr_prefix, seurat_obj, type="t"){
tcr <- read.csv(paste(tcr_prefix,"filtered_contig_annotations.csv", sep=""))
# Remove the -1 at the end of each barcode.(注意,此步骤如果标记使用不同的barcode,比如多了个-1,可以使用 tcr$barcode <- gsub("-1", "", tcr$barcode)进行提取)
# Subsets so only the first line of each barcode is kept,
# as each entry for given barcode will have same clonotype.
tcr <- tcr[!duplicated(tcr$barcode), ]
# Only keep the barcode and clonotype columns.
# We'll get additional clonotype info from the clonotype table.
tcr <- tcr[,c("barcode", "raw_clonotype_id")]
names(tcr)[names(tcr) == "raw_clonotype_id"] <- "clonotype_id"
# Clonotype-centric info.
clono <- read.csv(paste(tcr_prefix,"clonotypes.csv", sep=""))
# Slap the AA sequences onto our original table by clonotype_id.
tcr <- merge(tcr, clono[, c("clonotype_id", "cdr3s_aa")])
names(tcr)[names(tcr) == "cdr3s_aa"] <- "cdr3s_aa"
# Reorder so barcodes are first column and set them as rownames.
tcr <- tcr[, c(2,1,3)]
rownames(tcr) <- tcr[,1]
tcr[,1] <- NULL
colnames(tcr) <- paste(type, colnames(tcr), sep="_")
# Add to the Seurat object's metadata.
clono_seurat <- AddMetaData(object=seurat_obj, metadata=tcr)
return(clono_seurat)
}
s_balbc_pbmc <- add_clonotype("vdj_v1_mm_balbc_pbmc/vdj_v1_mm_balbc_pbmc_t_", s_balbc_pbmc, "t")
s_balbc_pbmc <- add_clonotype("vdj_v1_mm_balbc_pbmc/vdj_v1_mm_balbc_pbmc_b_", s_balbc_pbmc, "b")
head(s_balbc_pbmc[[]])我给解释一下以上function中每一步都在干什么:
首先读入contig_annotations.csv,并赋给tcr;
去除tcr中重复的barcode,即如果具有相同的barcode,将以第一次出现的barcode为主来去重;
将tcr中的barcode,raw_clonotype_id赋值于tcr;
读入clonotypes.csv,并赋给clono;
将tcr和colono进行merge(单细胞分析Seurat使用相关的10个问题答疑精选!),并赋给tcr;
将最后得到的,带有barcode,raw_clonotype_id和colono的tcr对象以metadata的形式加入seurat object中。
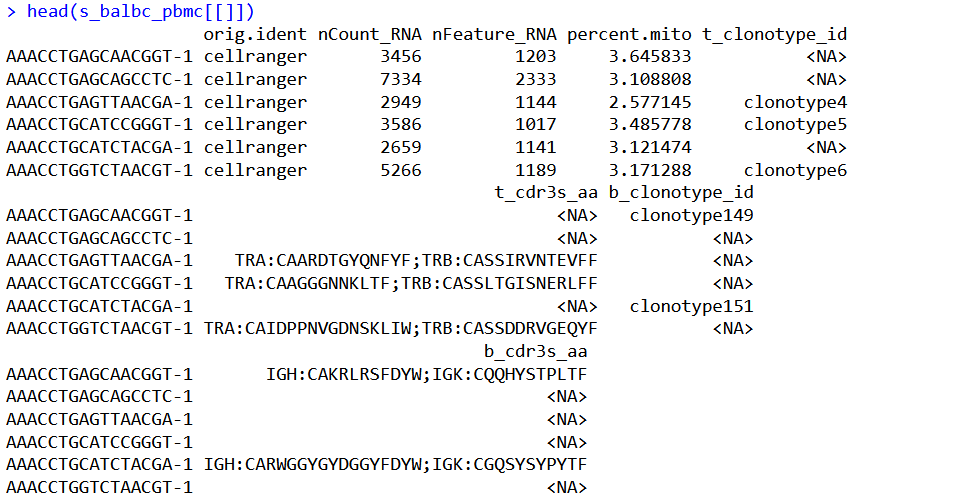
发现很多的NA,非常正常啊,不是每个细胞都是T/B cell,然后还列出来了T/B CDR3的蛋白序列。
table(!is.na(s_balbc_pbmc$t_clonotype_id),!is.na(s_balbc_pbmc$b_clonotype_id))

s_balbc_pbmc <- subset(s_balbc_pbmc, cells = colnames(s_balbc_pbmc)[!(!is.na(s_balbc_pbmc$t_clonotype_id) & !is.na(s_balbc_pbmc$b_clonotype_id))]) #删除同时表达T、B克隆表型的细胞 s_balbc_pbmc
进行常规workflow
s_balbc_pbmc <- subset(s_balbc_pbmc, percent.mito <= 10) s_balbc_pbmc <- subset(s_balbc_pbmc, nCount_RNA >= 500 & nCount_RNA <= 40000) s_balbc_pbmc <- NormalizeData(s_balbc_pbmc, normalization.method = "LogNormalize", scale.factor = 10000) s_balbc_pbmc <- FindVariableFeatures(s_balbc_pbmc, selection.method = "vst", nfeatures = 2000) all.genes <- rownames(s_balbc_pbmc) s_balbc_pbmc <- ScaleData(s_balbc_pbmc, features = all.genes) s_balbc_pbmc <- RunPCA(s_balbc_pbmc, features = VariableFeatures(object = s_balbc_pbmc))

use.pcs = 1:30 s_balbc_pbmc <- FindNeighbors(s_balbc_pbmc, dims = use.pcs) s_balbc_pbmc <- FindClusters(s_balbc_pbmc, resolution = c(0.5)) s_balbc_pbmc <- RunUMAP(s_balbc_pbmc, dims = use.pcs) DimPlot(s_balbc_pbmc, reduction = "umap", label = TRUE)
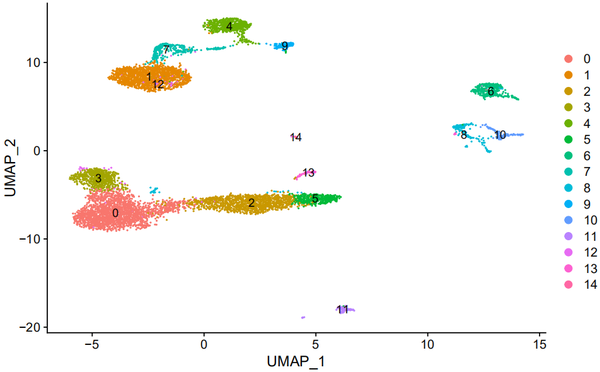
让我们看看T细胞的marker的表达情况:
t_cell_markers <- c("Cd3d","Cd3e")
FeaturePlot(s_balbc_pbmc, features = t_cell_markers)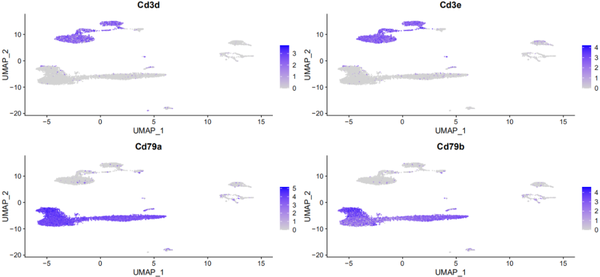
比如我知道某个b_cdr3s_aa我感觉特有意思,想在UMAP 图上进行表示,于是我先把他的蛋白序列找了出来,IGH:CARWGGYGYDGGYFDYW;IGK:CGQSYSYPYTF,然后:
DimPlot(s_balbc_pbmc, cells.highlight = Cells(subset(s_balbc_pbmc, subset = b_cdr3s_aa + == "IGH:CARWGGYGYDGGYFDYW;IGK:CGQSYSYPYTF")))
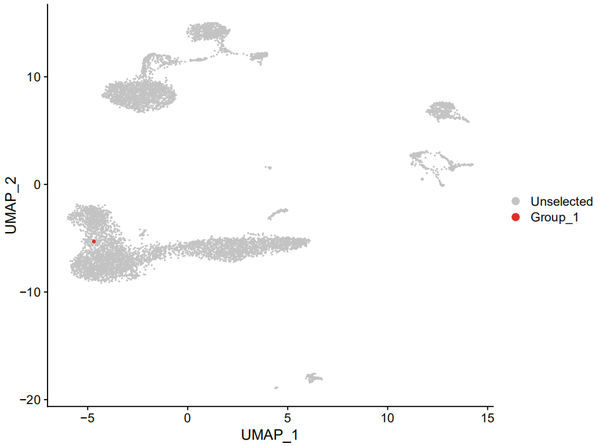
万“绿”丛中一点红!
当然你也可以在metadata中纳入更多的TCR/BCR中的有关信息,比如我们把annotation.csv中的chains也纳入进来的话,就是这个样子滴:
p2 <-DimPlot(s_balbc_pbmc,group.by = "t_chain") p2
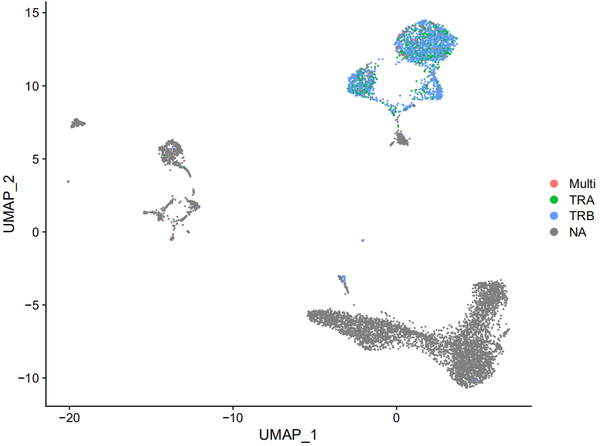
参考来源
https://github.com/ucdavis-bioinformatics-training/2020-Advanced_Single_Cell_RNA_Seq/blob/master/data_analysis/VDJ_Analysis_fixed.md
你可能还想看
https://wap.sciencenet.cn/blog-118204-1241281.html
上一篇:复现Cell附图 |类器官的单细胞分析
下一篇:NC文章详解 | 鼠成纤维细胞单细胞分析发现成纤维细胞在心肌细胞成熟中起关键作用