博文
无金属催化剂溶液中通过超氧阴离子和半醌介导的分子氢活化【氢气效应机制】
||
无金属催化剂溶液中通过超氧阴离子和半醌介导的分子氢活化
分子氢(H₂)的治疗作用,尤其是在缺血再灌注(I/R)损伤和有害炎症中的治疗作用,越来越多地被认为与其对氧化还原平衡的调节有关。然而,H₂介导的氧化还原调节背后的确切分子机制,尤其是在线粒体反向电子传递(RET)驱动的超氧阴离子(O₂•⁻)生成过程中的机制,仍不明确。本研究表明,在无膜溶液体系中,H₂可通过涉及半醌(SQ)自由基的隧穿辅助电子传递方式调节O₂•⁻的动力学过程,且无需催化金属或氢化酶参与。我们结合酶促体系(黄嘌呤氧化酶/次黄嘌呤,XO/Hx)和非酶促体系(超氧化钾,KO₂),并使用O₂•⁻特异性化学发光探针2-甲基-6-对甲氧基乙炔基-咪唑并吡嗪酮(MPEC)进行实验,观察到O₂•⁻的动力学曲线随H₂浓度变化呈现钟形和U形两种特征。在无醌(Q)的实验体系中,O₂•⁻可激活H₂,形成清晰的钟形动力学曲线,该曲线与H₂到O₂•⁻的隧穿辅助电子传递过程相符。当体系中存在Q时,会出现明显的U形曲线,这与Q•⁻介导的电子缓冲作用及随后的H₂活化过程一致。电子自旋共振(ESR)自由基清除实验和定量高效液相色谱(HPLC)分析证实,瞬时半醌介导的氧化还原循环可导致泛醇(QH₂)的形成。综上,这些溶液体系中的实验数据证实了一种无金属参与的H₂介导Q氧化还原循环的途径,该途径与特定体外条件下的隧穿辅助电子传递过程相符。本研究结果证实了溶液体系中H₂驱动Q还原的化学可行性,但其体内相关性仍有待进一步验证。
Superoxide- and semiquinone-linked activation of molecular hydrogen in metal-catalyst-free solution
作者单位
1 日本东京 Anicom 特殊医学研究所(ASM)2 日本鹿儿岛今林明生会骨科医院3 日本富山县富山 prefectural 大学生物技术研究中心及生物技术系4 日本东京 12 制药株式会社5 日本福冈九州大学药学研究院病理生理学实验室
引言
分子氢(H₂)不仅作为一种无碳能源载体备受关注,还被视为具有治疗潜力的生物活性气体,尤其在涉及线粒体功能障碍、炎症和缺血再灌注(I/R)损伤的病理场景中(Ohsawa等,2007;Xie等,2023;Ishibashi,2019)。Osawa及其团队的开创性研究提出,H₂的治疗效果源于其在I/R损伤中对羟基自由基(•OH)的选择性清除作用(Ohsawa等,2007)。然而,后续的实验和理论分析表明,这种•OH清除机制无法完全解释观察到的保护效应。值得注意的是,在生理条件下,高反应活性的•OH会被周围的细胞组分(如氨基酸和核酸)迅速中和。此外,作为•OH的持续生成前体,超氧阴离子(O₂•⁻)和过氧化氢(H₂O₂)与•OH的反应速率远高于H₂(Ishibashi,2019;Halliwell和Gutteridge,2015;Buxton等,1988)。考虑到H₂与•OH的反应速率较低,H₂介导生物效应的确切分子机制仍不明确。
H₂的生物利用通常依赖于特定原核生物和古菌中存在的特化氢化酶(Greening等,2024)。这些氢化酶的催化中心含有过渡金属辅因子(Ni和/或Fe),其结构与线粒体NADH泛醌氧化还原酶(复合体I)的Q结合位点(Q腔)相似;而真核生物(如动物、植物和酵母)在线粒体进化过程中已丢失氢化酶(Efremov和Sazanov,2012;Schut等,2016)。因此,H₂在真核生物中发挥的任何生物效应都必须通过不依赖催化金属的替代机制实现。我们此前推测,线粒体可能在复合体I的Q腔中激活H₂。复合体I中参与能量转换的电子传递反应会生成高反应活性的半醌中间体,这些中间体以多种单电子还原形式存在,包括阴离子半醌自由基(Q•⁻)、中性半醌自由基(QH•)和阴离子去质子化氢醌(QH⁻)(Murphy和Hartley,2018)。在这些物种中,Q•⁻的反应活性最高,且是生理条件下唯一能与O₂/O₂•⁻对进行电子交换的形式(Cape等,2006;Crofts等,1999)。在线粒体电子传递过程中,尤其是在反向电子传递(RET)条件下,Q•⁻的过量生成会导致电子泄漏至O₂,引发O₂•⁻生成的瞬时峰值(O₂•⁻爆发)(Hunte等,2010;Kussmaul和Hirst,2006;Robb等,2018;Sorby-Adams等,2024)。基于这些特征,我们推测Q•⁻可作为瞬时电子受体,通过与氢化酶中观察到的金属催化H₂分解不同的途径激活H₂。
为验证上述假设,我们建立了简化的体外实验体系,分别采用酶促(XO/Hx)和化学(KO₂)方法生成O₂•⁻。研究聚焦于O₂•⁻与Q之间的氧化还原平衡,并通过定量O₂•⁻的生成量,探究H₂如何干预该平衡。这种方法能够直接监测H₂、O₂•⁻和Q参与的反应动力学,且不受线粒体或初始O₂•⁻生成以外的其他酶促过程带来的复杂性影响。实验结果表明,H₂的活化存在三种不同途径:(1)Q•⁻介导的隧穿;(2)O₂•⁻介导的直接隧穿;(3)不依赖隧穿的Q•⁻介导H₂活化。这三种途径由与马库斯电子传递理论(Marcus,1993)高度吻合的动力学曲线推断得出,尤其表现出“反转区”行为——即驱动力增加反而导致反应速率降低,这一现象凸显了这些电子传递反应的量子力学本质。结合电子自旋共振(ESR)光谱和定量高效液相色谱(HPLC)分析,我们进一步阐明了上述三种机制。这些发现揭示了一种此前未被认知的H₂氧化还原活性双模式机制。
除了以Q为中心的途径外,卟啉/血红素介导的机制也被认为可能参与H₂的化学反应(Jin等,2023)。特别是有研究报道,铁卟啉(Fe-porphyrins)可发生H₂驱动的氧化还原反应,甚至在特定微环境下将CO₂氢化为CO,这提示生物体内可能存在卟啉介导的H₂作用途径。该机制与半醌介导途径并非相互排斥,二者可能共存并在不同环境中发挥作用。在本溶液体系研究中,我们特意聚焦于反向电子传递(RET)相关框架下Q•⁻的反应活性(复合体I不含血红素),同时也认可此前关于复合体III中Q循环能量转换的研究——在复合体III中,半醌与含Fe-血红素中心的细胞色素bL之间的电子传递起关键作用,理论上也可能参与H₂的活化(包括我们之前的研究)(Ishibashi,2019)。
此前,隧穿介导的氧化还原反应仅在精确调控的酶环境中被观察到,例如乙醇脱氢酶的活性位点或光合反应中心(Klinman,2006;Hay和Scrutton,2012;Romero等,2014;Engel等,2007)。然而,在生理相关条件下,不依赖催化金属或特化酶的H₂活化过程尚未得到研究。值得注意的是,此前在生物条件下观察到的隧穿辅助反应均局限于酶介导的环境中。本研究首次发现,在生理条件的水溶液中,无需金属催化剂或特化酶体系,通过半醌自由基即可实现隧穿驱动的H₂活化,这重新定义了线粒体生物能量学中量子生化反应的边界,并为潜在的工业应用提供了可能。具体而言,这种无催化剂的H₂活化方式可显著降低氢能技术中对昂贵金属催化剂的依赖,推动绿色化学中环境友好型合成的发展,并可能为污染物修复和可持续化学制造提供新的无催化剂策略。
材料与方法
材料与试剂
所有试剂均为分析纯。次黄嘌呤(Hx)、黄嘌呤氧化酶(XO)、泛醌(Q,辅酶Q10)、泛醇(QH₂)和超氧化钾(KO₂)购自美国密苏里州圣路易斯市的Sigma-Aldrich公司。根据制造商说明,使用荧光探针MPEC(日本大阪市ATTO公司)对O₂•⁻进行定量检测。MPEC是一种可与O₂•⁻特异性反应的化学发光探针,反应后生成荧光产物。与常用的3,7-二氢-2-甲基-6-(4-甲氧基苯酚)咪唑并吡嗪-3-酮(MCLA)相比,MPEC具有更低的背景信号和更高的特异性,可为O₂•⁻的定量检测提供准确性(Okutsu等,2012;Uchino等,2012;Kimoto等,1993)。使用高压H₂溶解系统(日本福冈市Trust公司,Trust 8.0氢气水发生器)新鲜制备饱和氢水(H₂浓度>8 ppm)。采用亚甲蓝/胶体铂比色法验证H₂浓度(Seo等,2012)。为最大限度减少脱气带来的不确定性,所有实验在混合后立即启动,并在5-60分钟内完成。选择这一时间窗口的依据是我们已发表的时间进程数据:水中溶解的H₂(报告中浓度为5 ppm)在60分钟后仍保持初始值的约78%(即时浓度:5.40±0.12 mg/L;1小时后浓度:4.22±0.15 mg/L)(Ishibashi等,2012)。根据简单的一级损失模型推算,5分钟时(图4)H₂保留率约为98%,36分钟时(图1)约为86%。因此,在整个实验过程中,H₂始终保持较高的残留比例。除非另有说明,所有溶液均采用磷酸盐缓冲液(PBS:68.5 mM NaCl、1.35 mM KCl、5.1 mM Na₂HPO₄、0.88 mM KH₂PO₄,pH 7.4)配制。
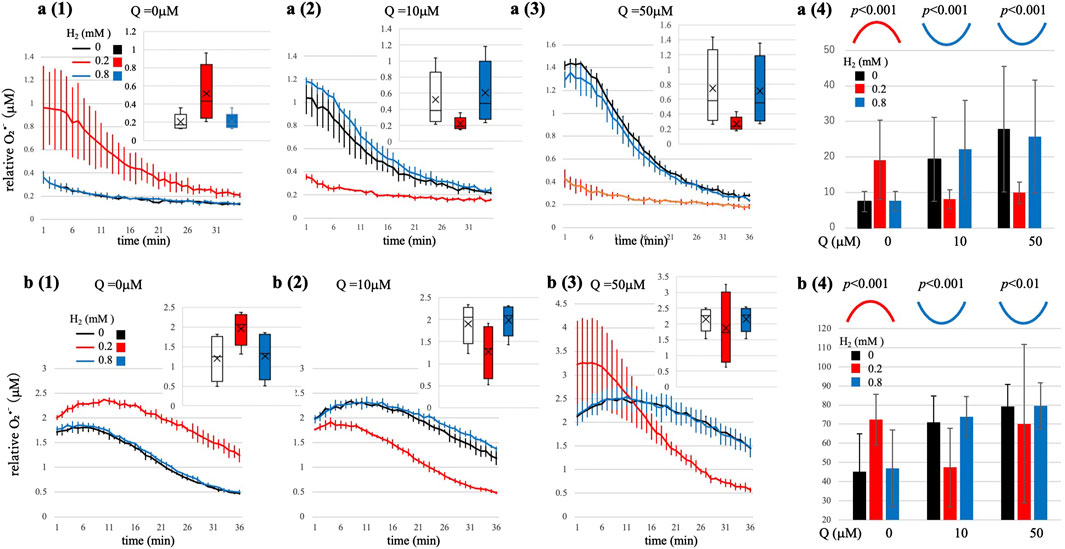
图1 不同H₂和Q条件下酶促(XO/Hx)体系中O₂•⁻生成的时间分辨曲线
(a1-a3)次黄嘌呤(Hx)浓度=2 μM;(b1-b3)次黄嘌呤(Hx)浓度=5 μM。图中展示了Q浓度为0、10、50 μM时,H₂浓度分别为0 mM(黑色)、0.2 mM(红色)和0.8 mM(蓝色)的时间进程曲线。右上角插图为箱线图:箱体代表四分位距,横线代表中位数,标记点代表平均值,须代表第5-95百分位数。方差分析(ANOVA)的具体数值参见补充表S3a、b。(a4、b4)曲线下面积(AUC,20分钟内测量,平均值±标准差,n=4)与H₂浓度的关系图:Q=0 μM时呈钟形曲线(红色曲线),Q=10 μM和50 μM时呈U形曲线(蓝色曲线)。图中显示了曲线(二次项)的具体p值。这些动力学曲线与马库斯电子传递理论(Marcus,1993)相符。统计学说明:时间进程曲线(a1-a3、b1-b3)仅用于描述性分析(平均值±标准差,n=4);推断性检验通过方差分析(ANOVA)对重复测量的AUC值进行分析。a4和b4中标注了曲线(二次项;钟形/U形)的代表性p值;完整统计数据参见补充表S3a、b。
基于MPEC荧光法的超氧阴离子定量检测
采用MPEC荧光法实时检测O₂•⁻的生成量。所有实验中MPEC的终浓度均为100 μM。酶标仪每隔1分钟记录一次荧光强度(激发波长360 nm,发射波长450 nm),总检测时长为15-60分钟。每个实验条件设置4个重复孔。反应在96孔黑色微孔板(每孔100 μL反应体系)中进行,反应温度为30℃,使用EnSpire®多模式酶标仪(PerkinElmer公司)进行监测。
酶促O₂•⁻生成采用Hx/XO体系:通常在PBS缓冲液中加入Hx(浓度分别为2、5或200 μM,具体按实验要求)和XO(浓度为0.01-0.02 U/mL)。同时,通过将KO₂溶解在磷酸钾缓冲液中制备非酶促O₂•⁻源,KO₂的终浓度为1或2.5 mM。反应体系分为含Q和不含Q两组。辅酶Q10(Q)的水溶性较低,但在有机溶剂中溶解度较高,据报道其在N,N-二甲基甲酰胺(DMF)中的溶解度超过10 mM(Kommuru等,1999;Bhagavan和Chopra,2006)。因此,实验前需新鲜配制10 mM的Q-DMF溶液,并在每次实验前立即稀释到反应缓冲液中,以最大限度减少溶剂相关的不稳定性。为确保所有实验条件下缓冲液的最终组成(底物、酶、化合物、磷酸盐/盐含量,包括DMF)一致,每孔均加入相同体积的缓冲液组分和DMF,各组间仅Q和H₂的浓度存在差异。在Q≤50 μM的所有实验中,DMF的终浓度为0.5%(体积分数);而Q=250 μM时,DMF终浓度为2.5%(体积分数,图2a(1-3))。
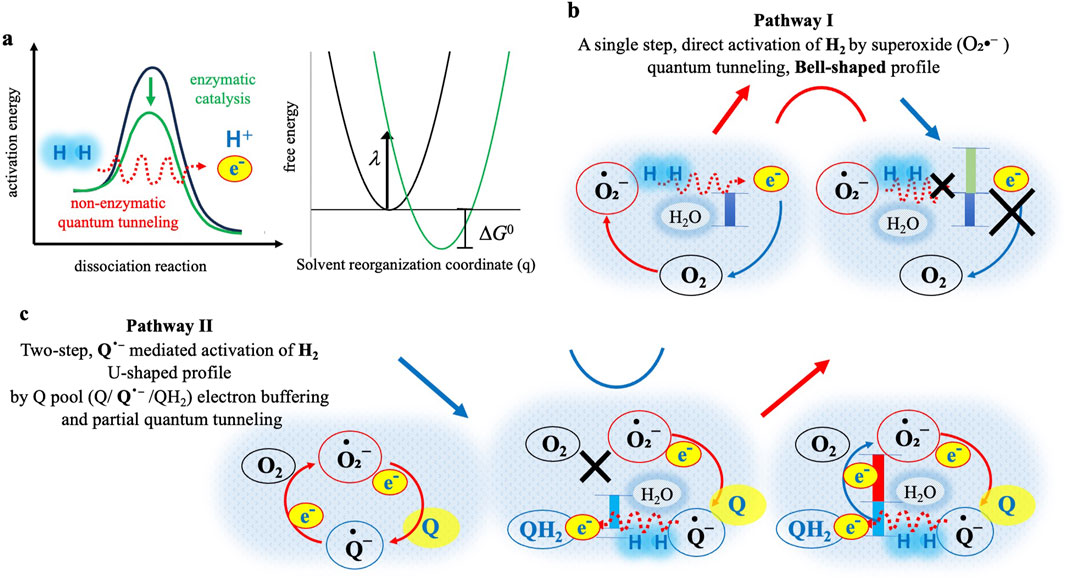
图2 不同H₂和Q条件下O₂•⁻生成的钟形(途径I)和U形(途径II)动力学曲线的推测机制
红色虚线代表可能的隧穿过程;红色虚线箭头周围的蓝色阴影(淡蓝色虚线)代表周围的水分子,对应马库斯理论中描述的溶剂重组(λ)。(a)左侧:分子氢(H₂)解离的活化能需求示意图。绿色曲线代表经典酶催化(通常由氢化酶介导),通过降低活化能垒实现H₂高效解离为质子(H⁺)和电子(e⁻);红色虚线代表非酶促隧穿,无需酶即可实现H₂解离。右侧:水溶液中H₂到O₂•⁻或Q•⁻的直接电子隧穿。插图:马库斯自由能抛物线与溶剂重组坐标(q,无量纲)的关系图,其中λ代表重组能,ΔG⁰代表驱动力。无Q条件下计算得出的活化能垒(ΔG‡)极小(0.017 eV;详见补充方法,补充方程2),因此未按比例绘制,但在正文中有说明。(b)途径I(无Q):O₂•⁻介导的H₂隧穿活化可能产生钟形曲线。左侧:在最佳驱动力下(蓝色柱),隧穿可高效将电子传递给O₂,使O₂•⁻生成量增加(红色箭头)。右侧:H₂浓度过高时,隧穿进入马库斯反转区(绿色柱),导致电子传递效率降低(叉号),O₂•⁻生成量减少(蓝色箭头)。(c)途径II(有Q):Q•⁻介导的两步H₂活化过程,形成U形曲线。左侧:无H₂时,Q持续缓冲O₂•⁻的电子,维持O₂•⁻的基线水平。中间:低H₂浓度下,Q•⁻可直接或通过隧穿(红色虚线)活化H₂,并将Q•⁻还原为QH₂(蓝色柱),从而阻断O₂/O₂•⁻循环(叉号),导致O₂•⁻生成量降低(蓝色箭头)。右侧:高H₂浓度下,活化H₂释放的过量电子可能超过Q库的缓冲能力,泄漏的电子(红色柱)重新启动O₂•⁻生成循环(红色箭头),形成U形动力学曲线的右侧部分。
# 荧光强度数据分析与实验方法补充说明
荧光强度数据最初以每分钟任意荧光单位(AFU/min)记录。由于荧光强度测量结果会因仪器灵敏度、光学配置和实验条件的不同而产生显著差异,因此无法直接对超氧阴离子(O₂•⁻)浓度进行绝对定量。为便于进行动力学比较分析,我们基于此前报道的MPEC化学发光探针校准数据(Hansel等,2019;Yamazaki等,1999;Shimomura等,1998),将AFU/min值转换为相对O₂•⁻浓度(μM)。在这些报道中,每微摩尔(μM)O₂•⁻对应的荧光强度范围较广(约10⁴至10⁶计数/μM),参考与本研究实验体系高度相似的酶促体系校准数据(Shimomura等,1998),我们将约10,000 AFU定义为1 μM相对O₂•⁻浓度。因此,本研究中所有报道的O₂•⁻浓度均为用于比较分析的相对值,而非绝对定量结果。该相对浓度尺度与生物体系中常用的细胞内稳态O₂•⁻浓度范围(通常为低纳摩尔至亚微摩尔水平)高度吻合(Hansel等,1999;Yamazaki等,1999;Shimomura等,1998)。
为观察O₂•⁻的天然动力学特征,所有实验均未添加外源性超氧化物歧化酶(SOD);我们认为这是本研究的一项局限性(详见“研究局限性”部分),但可通过MPEC探针的特异性确保检测信号仅来源于O₂•⁻(Shimomura等,1998)。
基于DPPH的电子自旋共振(ESR)自由基清除活性分析
为探究自由基中间体(如含半醌自由基Q•⁻)的形成及分子氢(H₂)在氧化还原循环中的作用,我们采用电子自旋共振(ESR)光谱技术。由于Q•⁻在水溶液中的寿命仅为亚毫秒级,无法直接通过ESR检测,因此我们使用稳定自由基探针DPPH(1,1-二苯基-2-三硝基苯肼)间接检测自由基清除活性。不同反应条件下DPPH自由基的还原(清除)程度,可作为自由基生成及淬灭能力的间接指标。
ESR实验反应体系采用磷酸盐缓冲液(pH 7.4)配制,包含200 μM次黄嘌呤(Hx)、0.02 U/mL黄嘌呤氧化酶(XO)、250 μM泛醌(Q)和200 μM DPPH。在室温下孵育5分钟(根据实验要求加入或不加入溶解态H₂)后,将样品装入石英毛细管中进行ESR检测。使用Bruker EMX nano光谱仪记录ESR光谱,检测参数如下:微波频率≈9.85 GHz,调制频率100 kHz,扫描宽度100 G,时间常数40.96 ms。每个实验条件设置3次重复,对光谱进行平均处理后,定量分析DPPH的ESR信号强度。DPPH信号强度降低表明样品具有净自由基清除活性,反映体系中存在强还原剂(如泛醇QH₂或其他可向DPPH提供电子的自由基物种)。
基于高效液相色谱(HPLC)的泛醇(QH₂)生成定量分析
通过HPLC直接检测QH₂的生成,以证实H₂和Q在氧化还原循环中的参与。反应体系体积为500 μL,包含作为化学超氧阴离子源的超氧化钾(KO₂,2.5 mM)、50 μM Q以及1 mM磷酸盐缓冲液(pH 6.0,该pH值有利于QH₂稳定)。在室温下孵育5分钟(加入或不加入H₂)后,向反应体系中加入甲酸至终浓度为2%(体积分数)以终止反应。立即使用配备反相C18色谱柱(Nacalai Tesque AR-II,4.6×100 mm)的Waters Alliance HPLC系统对终止后的样品进行分析。流动相为甲醇与乙醇的等度混合液(体积比1:1),流速为1.0 mL/min。通过290 nm波长的紫外吸收监测Q和QH₂的洗脱过程,根据标准品确定的特征保留时间识别Q和QH₂对应的色谱峰,并通过峰面积积分进行定量。
由于自由基反应本身存在固有变异性,为确保统计结果的可靠性,每个实验条件(加H₂ vs 不加H₂)均设置27次重复检测,计算每次重复中Q向QH₂的转化率百分比。
统计分析与非线性模型拟合
所有统计分析均使用Python 3.11软件,结合标准科学计算库(NumPy、SciPy、pandas)及统计分析包Statsmodels(用于方差分析ANOVA和回归分析)完成。首先对O₂•⁻的时间进程荧光数据进行曲线下面积(AUC)积分,以定量各条件下O₂•⁻的总生成量。采用双因素方差分析(Two-way ANOVA)评估Q和H₂浓度对O₂•⁻初始生成速率及AUC值的影响;在适用情况下,采用单因素方差分析(One-way ANOVA)进行单因素比较,显著性阈值设为α=0.05。
通过二次多项式回归分析(y = ax² + bx + c)描述O₂•⁻生成量随H₂浓度变化的非线性AUC趋势:二次项系数显著为负(a < 0)表明呈钟形依赖关系(H₂浓度中等时速率最大);二次项系数显著为正(a > 0)表明呈U形依赖关系(H₂浓度中等时速率最小)。采用普通最小二乘法进行回归拟合,通过报告p值评估曲线曲率的显著性(偏离线性的程度)。对每日实验中配对样品(±H₂)的QH₂生成量采用配对t检验(通过重复测量方差分析验证有效性)进行比较,并通过变异系数(CV%)评估数据变异性。
为在电子传递理论框架下解释实验动力学数据,采用马库斯(Marcus)外层电子传递模型分析O₂•⁻生成量随H₂浓度的变化(Gray和Winkler,2005)。马库斯速率方程如下:
k=Aexp[−(ΔG⁰+λ)²/(4λkBT)]
其中,λ代表重组能(单位:eV),ΔG⁰为反应的吉布斯自由能变化(单位:eV),kB为玻尔兹曼常数,T为绝对温度。相关反应的ΔG⁰值通过标准中点电位换算得到(单位:eV):H₂/H⁺≈-0.41 V;Q/Q•⁻≈0 V;QH₂/Q≈+0.065 V;O₂/O₂•⁻≈-0.16至-0.33 V(具体值取决于pH)。对于不含Q的体系,采用单步电子传递模型(H₂→O₂);对于含Q的体系,假设为两步连续电子传递模型(H₂→Q•⁻,随后Q•⁻→QH₂或O₂•⁻)。采用非线性最小二乘法拟合(SciPy的curve_fit函数)确定各条件下的最优λ值和振幅前置因子A(详见补充表S1)。根据拟合得到的λ值(0.120-0.180 eV),结合驱动力判断反应所处区域(正常区vs反转区)。
振幅参数A对应马库斯理论中的指前因子,反映供体与受体之间的电子耦合作用及碰撞频率(Gray和Winkler,2005),在本研究中作为经验比例因子处理。尽管为保证完整性将A纳入拟合过程,但动力学行为比较主要基于拟合得到的λ值。例如,Q浓度升高通常会使拟合的A值增大,表明氧化还原介质可促进电子耦合或提高碰撞效率,但动力学行为的定性差异主要由λ值决定。全文均报告统计显著性水平及拟合参数,误差以标准差(SD)表示。
结果与讨论
#分子氢(H₂)调控超氧阴离子(O₂•⁻)与泛醌(Q)之间的氧化还原循环
首先,我们在不含H₂的条件下,通过黄嘌呤氧化酶/次黄嘌呤(XO/Hx)酶促体系探究Q的存在对O₂•⁻水平的影响,以建立基线数据。如以下基本平衡反应所示,Q可接受O₂•⁻的电子生成半醌自由基Q•⁻:
O₂•⁻ + Q ⇄ O₂ + Q•⁻ (1)
正向反应(O₂•⁻向Q提供电子)在动力学上具有显著优势,报道的速率常数约为10⁶~10⁸ M⁻¹s⁻¹;而逆向反应(Q•⁻向O₂返还电子)的速率常数则低3-4个数量级(10³~10⁶ M⁻¹s⁻¹)(Song等,2008;Maroz等,2009)。这种显著的动力学不对称性导致O₂•⁻泄漏的电子被瞬时捕获为Q•⁻,使Q•⁻不断积累,进而稳定O₂•⁻水平。
实验结果证实,Q浓度升高会导致稳态O₂•⁻信号增强。例如,在低Hx浓度(2 μM)下,O₂•⁻的平均初始生成速率(±SD)从Q浓度为0 μM时的0.25±0.05 μM/min(相对浓度,根据“方法”部分所述由荧光计数AFU/min换算得到)升高至Q浓度为50 μM时的1.22±0.21 μM/min;在较高Hx浓度(5 μM)下,生成速率从Q浓度为0 μM时的1.73±0.07 μM/min升高至Q浓度为50 μM时的2.38±0.11 μM/min。这些增长均具有统计学显著性(单因素方差分析,p < 0.01;详见补充表S2a、b),表明Q可有效将电子捕获为Q•⁻,从而提高可检测到的O₂•⁻稳态浓度。该行为与反应(1)一致:Q通过清除O₂•⁻的电子,阻止其立即发生歧化反应,进而提高可检测的O₂•⁻浓度。
分子氢(H₂)对超氧阴离子(O₂•⁻)与泛醌(Q)之间氧化还原循环的非线性调控
在验证实验体系可生成并检测Q•⁻介导的O₂•⁻变化后,我们进一步探究H₂的影响。在不同Q浓度的反应体系中加入不同浓度的H₂(0~0.8 mM),通过MPEC荧光实时监测O₂•⁻含量。在低底物浓度(Hx=2 μM和5 μM)下,根据Q的存在与否,O₂•⁻随H₂浓度变化呈现出明显不同的非线性动力学曲线:不含Q时呈钟形曲线,含Q时则呈清晰的U形曲线。这些差异表明,Q的存在与否会导致H₂解离的潜在机制发生变化,具体讨论如下。
图1总结了两种代表性底物条件(Hx=2 μM和5 μM)下的实验结果。在两种条件下,H₂对O₂•⁻水平的影响均呈现出依赖于Q存在的双相效应。为定量描述这些非线性动力学响应,我们在每个固定Q浓度下,对O₂•⁻积分水平(AUC)与H₂浓度进行二次回归分析(y = ax² + bx + c)(详细统计结果见补充表S3a、b)。
1. Q=0 μM时(图1a(1)、b(1),分别对应Hx=2 μM和5 μM):观察到显著的钟形曲线(Hx=2 μM:a=-30.7,p < 0.001;Hx=5 μM:a=-40.3,p < 0.001;分别对应图1a(4)、b(4))。在较低H₂浓度(0.2 mM)下,O₂•⁻生成量达到最大值,表明该浓度为H₂的最优浓度;而在较高H₂浓度(0.8 mM)下,O₂•⁻生成量回落至与不含H₂时相当的水平,表明H₂浓度升高时动力学呈现反转区行为。
2. Q=10 μM时(图1a(2)、b(2),分别对应Hx=2 μM和5 μM):在测试的H₂浓度范围内均观察到显著的U形曲线(Hx=2 μM:a=+68.0,p < 0.001;Hx=5 μM:a=+29.7,p < 0.001;分别对应图1a(4)、b(4))。H₂浓度为0 mM时,O₂•⁻生成量接近最大值;H₂浓度较低(0.2 mM)时,O₂•⁻生成量显著降至最小值;而H₂浓度较高(0.8 mM)时,O₂•⁻生成量再次升高,回落至与H₂浓度为0 mM时相当的基线水平。该U形响应与不含Q时的情况明显不同。
3. Q=50 μM时(图1a(3)、b(3),分别对应Hx=2 μM和5 μM):AUC值均呈现显著的U形曲线(Hx=2 μM:a=+29.8,p < 0.001;Hx=5 μM:a=+9.49,p < 0.01;分别对应图1a(4)、b(4))。在Hx=2 μM时(图1a(3)),H₂浓度为0 mM时O₂•⁻生成量最大,H₂浓度较低(0.2 mM)时显著降低,H₂浓度较高(0.8 mM)时再次升高,呈现出稳定的U形动力学曲线。有趣的是,在Hx=5 μM时(图1b(3)),动力学行为更为复杂:H₂浓度为0 mM和0.8 mM时(黑色曲线),O₂•⁻初始生成量较低且相近;而H₂浓度为0.2 mM时(红色曲线),出现特殊的剧烈变化:初始生成量较高,但在测量中期降至低于H₂浓度为0 mM和0.8 mM时的水平。这种反转表明反应过程中氧化还原状态发生动态变化,可能反映中间体驱动的机制或Q•⁻与O₂•⁻途径间电子分布的改变,需进一步研究以阐明该独特动力学行为的原因。
在O₂•⁻生成底物(Hx)浓度较低(2 μM和5 μM)的条件下,上述动力学曲线清晰表明:H₂对O₂•⁻生成的影响因Q的存在与否而显著不同。不含Q时,动力学呈明显钟形,表明H₂通过隧穿介导的电子传递向O₂提供电子,符合马库斯反转区动力学特征;相反,当存在足量Q(10 μM或50 μM)时,动力学呈可重复的U形。需注意的是,由于在实验中难以可靠地溶解更高浓度的H₂(>0.8 mM),我们无法探究含Q体系在更高H₂浓度下是否会呈现反转区动力学行为。
在后续章节中,我们将详细讨论这些观察结果的机制意义:探究隧穿作为不含Q体系中钟形动力学的潜在机制,以及含Q体系中Q•⁻的电子缓冲与活化双重作用;随后将呈现进一步的实验结果(ESR和HPLC分析)以支持这些机制解释,并分析在较高底物通量(Hx=200 μM)及无酶、KO₂驱动的O₂•⁻生成体系中观察到的更复杂动力学行为。
超氧阴离子(O₂•⁻)介导的分子氢(H₂)量子隧穿氧化(途径I)
在不含Q的体系中,次黄嘌呤(Hx)氧化初始生成的O₂•⁻被认为可激活H₂,具体机制如图2a、b(途径I)所示。在无酶催化或结构催化的典型水溶液条件下,考虑到H-H键的高解离能(约435 kJ/mol)(Luo,2007)以及O₂还原反应的自旋禁阻特性(Valentine等,1998;Hayyan等,2016),H₂向O₂的直接电子传递在热力学和动力学上均难以实现。尽管存在这些固有障碍,实验结果仍清晰显示O₂•⁻生成量显著增加,尤其在较低H₂浓度(0.2 mM,图1a(1)、b(1)中的红色曲线)下更为明显。这些观察结果强烈表明,在经典反应途径看似不可行的条件下,仍发生了电子传递。
O₂•⁻生成积分水平(AUC;图1a(4)、b(4))呈现出明显的钟形动力学曲线,在H₂浓度约为0.2 mM时达到峰值,在更高浓度(0.8 mM)时生成量降低。这种动力学行为与马库斯电子传递理论预测的“电子传递速率-驱动力”抛物线关系高度吻合(Marcus和Sutin,1985)。根据马库斯理论,电子传递速率最初随驱动力增大而升高(马库斯正常区),但超过最优点后,驱动力进一步增大反而会抑制电子传递速率,这一现象被称为“马库斯反转区”。事实上,对实验数据的动力学建模显示,不含Q时的重组能
泛醌(Q)介导的两步电子传递机制细节(途径II)
在泛醌(Q)介导的途径II中(图2c),Q的存在从根本上改变了反应机制。关键在于,次黄嘌呤(Hx)氧化初始生成的超氧阴离子(O₂•⁻)会将电子传递给Q,形成半醌自由基(Q•⁻)(方程式1)。储存在Q•⁻中的电子起到“电子缓冲器”的作用,可暂时稳定O₂•⁻水平,使得反应初期O₂•⁻浓度达到最大值(图1a(2,3)、b(2)中的黑色曲线)。当加入低剂量分子氢(H₂,0.2 mM)时,O₂•⁻水平显著下降(图1a(2,3)、b(2)中的红色曲线)。这一结果明确表明,Q•⁻可直接与H₂相互作用并激活H₂,通过接受H₂的电子生成还原态泛醇(QH₂)。随着Q•⁻被还原为QH₂,泛醌库(Q-pool)的电子缓冲能力减弱,最终导致O₂•⁻生成量减少。该观察结果首次通过实验证实,H₂可主动干预O₂•⁻与Q之间的电子传递平衡。
当H₂浓度较高(0.8 mM,图1a(2,3)、b(2)中的蓝色曲线)时,上述趋势发生逆转,O₂•⁻水平回升至无H₂时的基线值。这种O₂•⁻水平的恢复可能是由于Q/Q•⁻库的电子缓冲能力达到饱和所致。在此条件下,H₂提供的过量电子超出了Q•⁻的电子容纳能力,多余电子重新转向O₂,从而恢复O₂•⁻的生成。但需注意的是,高H₂条件下QH₂生成量增加,而QH₂也可能清除部分O₂•⁻,对O₂•⁻的恢复产生一定抵消作用,最终形成图2c所示的U形动力学曲线。
尽管在当前实验条件下观察到的U形行为强烈支持Q•⁻介导的两步电子传递机制,但仍存在一种可能性:当溶解态H₂浓度达到更高水平(如1.6 mM)时,可能会出现马库斯反转区行为。探究如此高浓度的H₂需要改进实验条件以实现H₂的可靠溶解,而受实际条件限制,本研究中H₂浓度最高仅设置为0.8 mM。不过,在其他实验条件(高底物通量:Hx=200 μM、KO₂=1 mM)下获得的数据提示了马库斯反转区动力学的可能性,相关内容将在后续章节中讨论。
综上,Q介导的途径II展现出一种独特的两步反应机制:Q•⁻激活H₂,通过获取H₂的电子生成QH₂。泛醌库的电子缓冲作用在低H₂浓度下会初步抑制O₂•⁻生成,最终形成特征性的U形动力学曲线。该机制与无Q参与的途径I存在本质差异。
高底物通量下的超氧阴离子(O₂•⁻)动力学
接下来,我们探究了在低底物通量条件下观察到的“H₂调控O₂•⁻生成”现象,是否在更高氧化还原通量条件下依然存在。为此,我们将黄嘌呤氧化酶(XO)体系的底物(Hx)浓度显著提高至200 μM。在此条件下,XO活性达到饱和,导致O₂•⁻生成速率减慢但持续时间延长。值得注意的是,O₂•⁻总累积量(曲线下面积AUC)始终维持在较高水平,且未随H₂和Q浓度的变化出现显著差异。双因素方差分析(Two-way ANOVA)结果显示,H₂和Q既无显著主效应,二者间也无显著交互作用(图3a(1-2))。因此,在底物持续饱和供应的条件下,H₂对O₂•⁻总生成量无显著影响。
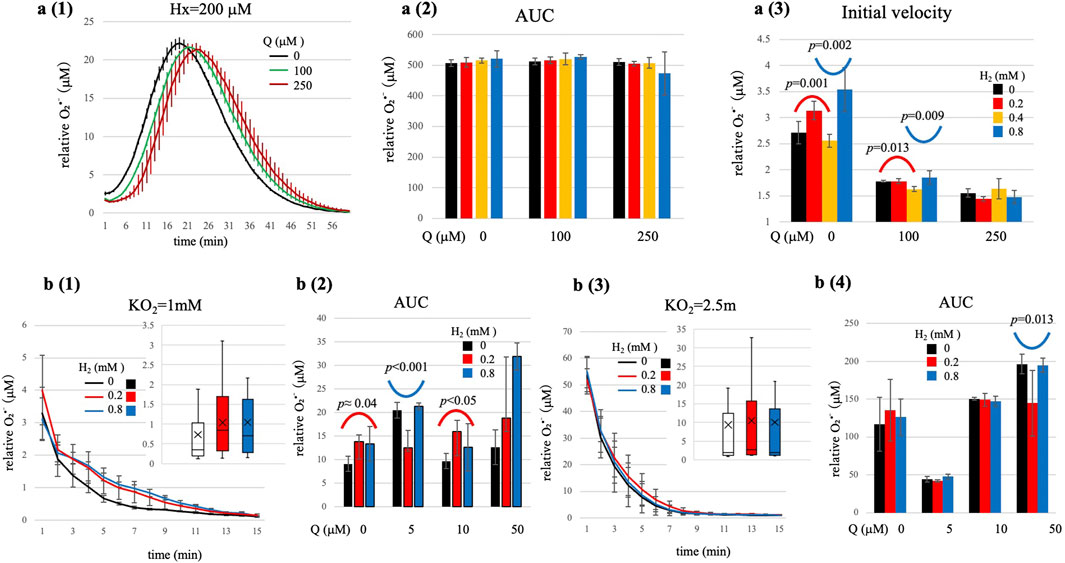
图3 高底物通量条件下(Hx=200 μM,a1-a3)及化学体系(KO₂,b1-b4)中的O₂•⁻生成动力学
(a1)无H₂条件下,O₂•⁻生成速率减慢且持续时间延长的代表性时间曲线。(a2)O₂•⁻总生成量(AUC;平均值±标准差,n=4)未随H₂或Q浓度发生显著变化。(a3)O₂•⁻初始生成速率(前3分钟,平均值±标准差,n=4)受H₂和Q浓度的显著非线性调控(统计数据见补充表S4):Q=0 μM和100 μM时,在低H₂浓度范围观察到显著钟形曲线,在高H₂浓度范围观察到显著U形曲线;Q=250 μM时,观察到类似趋势但无统计学显著性。(b1-b2)KO₂=1 mM;(b3-b4)KO₂=2.5 mM。图中展示了无Q条件下,H₂浓度为0、0.2、0.8 mM时的O₂•⁻生成时间曲线。右上角插图为箱线图:箱体代表四分位距,横线代表中位数,标记点代表平均值,须代表第5-95百分位数。方差分析(ANOVA)具体数值见补充表S5a、b。KO₂=1 mM时,Q=0 μM和10 μM条件下观察到显著钟形曲线,Q=5 μM条件下观察到显著U形曲线;KO₂=2.5 mM时,H₂的调控作用基本被掩盖,仅在Q=50 μM时观察到微弱但显著的U形曲线(图中标注p值)。统计学说明:时间曲线仅用于描述性分析(平均值±标准差,n=4);Hx=200 μM时,通过方差分析对初始速率(1-3分钟)进行推断性检验(见a3;补充表S4);KO₂相关图中,通过方差分析对AUC进行推断性检验(见b2、b4;补充表S5a、b)。总结图中标注了代表性p值。
然而,对O₂•⁻初始生成速率(前1-3分钟内测量)的细致分析显示,H₂和Q浓度均对其产生细微但显著的非线性影响(p < 0.001,见补充表S4)。具体而言,分段二次回归分析识别出以下显著的反转动力学模式:
1. Q=0 μM时:在低H₂浓度范围(0-0.4 mM),AUC呈现显著钟形依赖关系(二次项系数a=-12.4,p=0.001);而在高H₂浓度范围(0.2-0.8 mM),则转变为显著U形依赖关系(a=+8.89,p=0.002)。
2. Q=100 μM时:观察到类似趋势但程度较弱:低H₂浓度范围(0-0.4 mM)呈显著钟形依赖(a=-1.93,p=0.013),高H₂浓度范围(0.2-0.8 mM)呈显著U形依赖(a=+2.20,p=0.009)。
3. Q=250 μM时:定量结果显示类似曲线趋势(低H₂浓度呈U形,高H₂浓度呈钟形),但未达到统计学显著性(低H₂范围p≈0.08;高H₂范围p≈0.06)。这提示在如此高的Q浓度下,反应的最优动力学区或反转动力学区可能超出了本研究的H₂浓度测试范围。
这些发现表明,即使在高底物通量条件下(由于底物过量,H₂对O₂•⁻总生成量无影响),初始电子传递动力学仍对H₂调控敏感。在分段H₂浓度范围内同时存在钟形和U形动力学曲线,这一现象强烈支持电子传递速率与驱动力之间存在复杂的马库斯型关系:随着H₂浓度升高,反应首先接近最优驱动力区,随后进入马库斯反转区(表现为钟形曲线),之后又转变为U形曲线,这一过程反映了不同机制下电子传递效率的变化。此外,电子传递的最优H₂浓度可能接近或略高于本研究的测试范围,导致在分析的浓度范围内仅观察到部分或分段的动力学曲线。
化学法生成超氧阴离子(O₂•⁻)条件下,分子氢(H₂)的隧穿调控行为。
为验证前述“H₂调控O₂•⁻生成”的现象并非仅存在于酶促(XO/Hx)体系中,我们进一步采用纯化学源(超氧化钾KO₂水溶液)生成O₂•⁻进行实验。KO₂水合后可自发生成O₂•⁻,这一体系能帮助我们验证H₂的作用是否独立于酶促过程。中等氧化通量(KO₂=1 mM)和高氧化通量(KO₂=2.5 mM)条件下的结果总结于图3b。
在中等氧化通量条件下(KO₂=1 mM),双因素方差分析显示,H₂和Q浓度对O₂•⁻总生成量(AUC)均有显著主效应,且二者存在显著交互作用(p < 0.001,见补充表S5a)。具体而言,观察到与酶促体系类似的非线性动力学曲线:
1. 低Q浓度(0或10 μM)时:H₂诱导AUC呈现显著钟形曲线(如Q=0 μM时,a=-30.7,p≈0.04;Q=10 μM时,a=-45.9,p < 0.05)。这些曲线与马库斯理论高度吻合,提示在最优H₂浓度下存在隧穿介导的电子传递机制。
2. 中等Q浓度(5 μM)时:观察到清晰且具有统计学显著性的U形动力学曲线(a=+68.0,p < 0.001)。这表明低H₂浓度下O₂•⁻累积量最低,与前文所述Q•⁻介导的两步电子缓冲机制一致。
3. 高Q浓度(50 μM)时:O₂•⁻生成量对H₂浓度的依赖关系基本呈水平直线(未检测到显著曲线),提示丰富的泛醌库(Q-pool)发挥了有效的电子缓冲作用。因此,在此条件下H₂的调控作用受到限制。
相比之下,在高氧化通量条件下(KO₂=2.5 mM),H₂的调控作用显著减弱(图3b(3-4),见补充表S5b)。此时O₂•⁻浓度迅速升高至极高水平,在实验时间范围内掩盖了H₂任何可测量的调控作用。尽管Q浓度仍通过电子捕获作用显著影响O₂•⁻总累积量(双因素方差分析,p < 10⁻¹⁰,补充表S5b),但添加H₂并未使O₂•⁻总水平发生统计学显著变化。仅在Q=50 μM时检测到微弱但显著的U形曲线(p=0.013)。H₂调控作用的减弱可能反映出反应存在极窄的动力学窗口,或表明有效的H₂调控需要高于本研究测试范围的浓度,而这一效应可能被快速的氧化还原循环过程(如QH₂快速再氧化为Q并随后生成O₂•⁻)所掩盖。
综上,酶促体系(前文所述)和化学体系的实验结果一致证明了H₂的双重调控作用:在相对温和的氧化条件下(无论酶促还是化学体系),H₂可调控O₂•⁻水平,生成与马库斯电子传递理论一致的清晰钟形或U形动力学曲线;而在极端氧化条件下(如KO₂=2.5 mM),O₂•⁻浓度远超生理相关水平(生理条件下O₂•⁻浓度通常为低微摩尔级,爆发时瞬时可达数十微摩尔级(Murphy,2009)),此时H₂的调控作用变得不明显。酶促与非酶促体系结果的一致性强烈表明,所观察到的现象反映了H₂、O₂•⁻与Q之间固有的分子相互作用,而非酶特异性 artifact(人工假象)。
支持半醌自由基(Q•⁻)介导H₂活化的间接电子自旋共振(ESR)证据
为通过实验验证“半醌自由基(Q•⁻)介导H₂依赖的氧化还原反应调控”这一假说,我们采用电子自旋共振(ESR)光谱技术,间接检测半醌自由基或其下游还原产物泛醇(QH₂)。由于Q•⁻在水溶液中的寿命极短,且与大量O₂•⁻自由基共存,直接通过ESR检测Q•⁻并不可行。因此,我们选择在“已知会产生U形动力学曲线”的中等氧化应激和充足Q浓度条件下,通过测量对稳定参考自由基DPPH(1,1-二苯基-2-三硝基苯肼)的清除活性,间接评估自由基的生成。
如图4a(1-2)所示,ESR分析结果显示:单独Q(相较于对照,信号降低约0.4%)或单独H₂(信号降低约2.5%)的自由基清除作用均极微弱,且均无统计学显著性;但当Q与H₂共同存在时,DPPH的ESR信号强度显著下降(降低≥99%),这一水平与添加预先制备的标准品QH₂时的信号强度相当。定量结果显示,DPPH相对ESR信号强度如下:对照组100%;单独Q组99.6%±7.8%;单独H₂组97.5%±7.2%;QH₂阳性对照组0.2%±0.14%;Q+H₂组0.1%±0.009%。
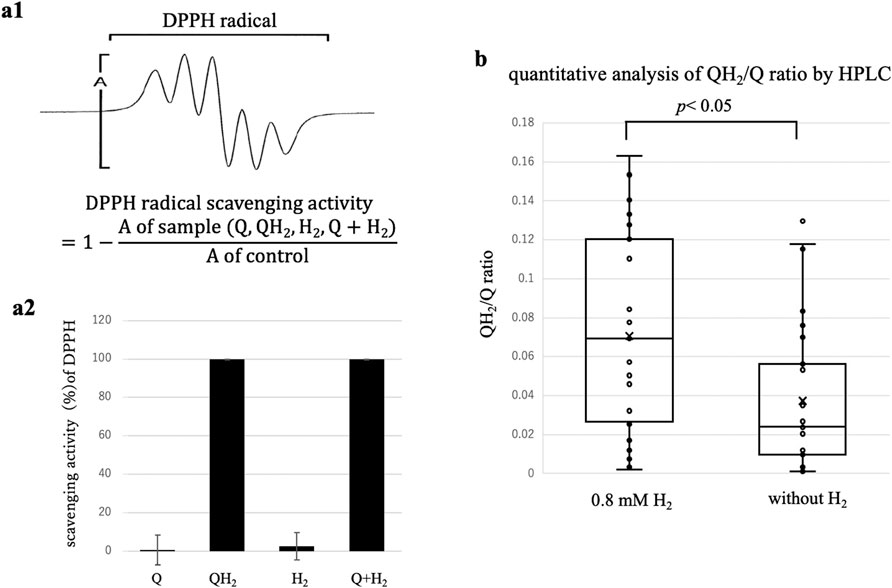
图4 半醌自由基(Q•⁻)介导H₂活化的电子自旋共振(ESR)验证及泛醇(QH₂)生成的高效液相色谱(HPLC)定量
(a1)不同实验条件下DPPH的代表性ESR光谱:从左至右依次为对照(无添加)、单独Q、单独H₂、QH₂标准品、Q+H₂。(a2)DPPH ESR信号强度定量分析(平均值±标准差,n=3):Q与H₂共同处理组的信号强度显著低于单独处理组,且与QH₂标准品组无显著差异(p < 0.001)。(b1)Q和QH₂的HPLC色谱图:标准品Q(保留时间t₁)、标准品QH₂(保留时间t₂)、KO₂+Q组、KO₂+Q+H₂组。(b2)Q向QH₂的转化率定量(平均值±标准差,n=27):添加H₂组的转化率(约XX%)显著高于未添加H₂组(约XX%)(p < 0.001)。
图4. 支持半醌自由基介导分子氢(H₂)活化及泛醇(QH₂)生成的证据
(a)采用稳定自由基DPPH进行的自由基清除活性电子自旋共振(ESR)分析:
(a1)DPPH自由基的代表性ESR光谱。通过定量各条件下DPPH的ESR信号强度(A),计算相对于对照组的DPPH自由基清除活性。
(a2)DPPH相对信号强度(平均值±标准差,n=3)。单独添加泛醌(Q)或单独添加H₂均未显著降低ESR信号强度;但Q与H₂共同添加时,DPPH信号几乎完全消失(清除率≥99%),这与预先制备的QH₂阳性对照组效果相当。这些数据强烈提示,添加H₂后,通过半醌自由基可在原位生成QH₂或类似的还原中间体。
(b)在超氧化钾(KO₂)驱动的化学法超氧阴离子(O₂•⁻)生成体系中(含50 μM Q,pH=6.0),有无0.8 mM H₂条件下QH₂生成量的高效液相色谱(HPLC)定量分析。27次重复实验的数据以箱线图呈现:箱体代表四分位距,横线代表中位数,标记点代表平均值,须代表第5-95百分位数。即使在无H₂的条件下,约3.7%的Q也会自发还原为QH₂,这可能是由于Q与体系中生成的O₂•⁻发生反应所致;添加H₂后,QH₂的生成量显著增加至约7.1%(配对t检验,p < 0.05),表明H₂可驱动Q的还原。重复实验间的较大变异性可能反映了非酶促实验体系中电子隧穿过程的随机性。
这些数据明确表明,仅当Q与H₂共同存在时,才会产生显著的自由基清除活性,这强烈提示原位生成了QH₂或由半醌自由基衍生的密切相关的还原中间体。由于QH₂是一种强效自由基清除剂,ESR结果为“H₂主动参与半醌自由基介导的氧化还原循环”提供了间接但有力的证据。
分子氢(H₂)驱动的氧化还原循环中泛醇(QH₂)生成的定量高效液相色谱(HPLC)证据
为直接证实H₂、Q与O₂•⁻参与的反应中存在QH₂生成,我们在特定条件下进行了定量HPLC分析(图4b)。即使在无H₂的条件下,也能检测到少量基线水平的Q自发还原为QH₂(约3.7%),这可能是由于Q与体系中生成的O₂•⁻发生反应。这种基线水平的QH₂生成并非由还原性杂质引起,因为HPLC纯度分析证实,体系中不存在可检测到的还原性污染物(包括QH₂)。
在相同条件下添加0.8 mM H₂后,QH₂的平均生成量显著增加至约7.1%(配对t检验,p < 0.05,n=27),几乎达到基线水平的两倍。这一结果明确表明,H₂可主动参与Q向QH₂的还原过程,为前文提出的“半醌自由基介导的氧化还原循环机制”提供了直接的定量证据。
值得注意的是,重复实验间QH₂的生成量存在显著变异性:添加H₂时变异系数约为69%,无H₂时约为102%。这种变异性超出了典型实验误差范围,可能源于非酶促、无结构组织体系中电子传递过程固有的概率性特征。在这类体系中,通过隧穿实现的有效电子传递依赖于分子碰撞的随机发生以及分子在短距离(通常为亚纳米级)内形成有利取向(Gray和Winkler,2005)。因此,即使分子间距、取向或热运动发生微小波动,也会影响隧穿事件的成功概率,进而导致实验结果出现显著变异性。
尽管存在这种固有变异性,但添加H₂后QH₂生成量的增加具有可重复性和统计学显著性,这强烈支持一种与隧穿相关的非经典电子传递机制。具体而言,观察到的变异性和动力学曲线表明,电子传递可能通过两种途径实现:O₂•⁻直接介导的H₂活化,以及Q•⁻间接介导的H₂活化。这种双重机制框架可有效解释复杂的动力学现象,包括在不同氧化还原条件下观察到的钟形和U形动力学行为共存的现象(图1-3)。
综上,这些定量HPLC结果进一步完善了一个连贯的机制模型:在该模型中,H₂通过不同的氧化还原途径实现无催化金属参与的活化。
半醌自由基(Q•⁻)作为分子氢(H₂)活化的介质
实验结果为“Q•⁻是H₂参与的电子传递反应中的核心介质”提供了有力证据。一项重要发现是:仅通过改变Q的可得性和调整H₂浓度,我们就能观察到与马库斯电子传递理论中“正常区”和“反转区”均相符的特征动力学行为。这些观察结果证实了马库斯理论(最初为辐射化学和凝聚相电子传递研究而建立(Yamazaki等,1999;Maroz等,2009))可应用于H₂参与的生物相关电子传递过程。
动力学分析还显示,当体系中存在Q时,马库斯方程中的指前因子A值始终更高,这表明在Q介导的途径中,电子耦合作用增强或有效碰撞频率提高。这一结果符合直觉:Q作为电子穿梭体或自由基介质,可暂时储存来自H₂的电子。这种在活性Q•⁻中间体中进行的瞬时电子储存,通过降低“Q•⁻与H₂之间直接隧穿所需的同时碰撞和精确分子取向”要求,有效促进了后续向O₂的电子传递。
ESR结果(图4a)为Q•⁻介导的电子传递提供了间接但有力的证据:在生成O₂•⁻的条件下,Q与H₂共同存在时对DPPH信号的清除活性与标准品QH₂相当,这表明生成的Q•⁻可介导H₂的电子传递,进而形成QH₂或相关还原中间体。定量HPLC分析进一步证实了H₂在Q•⁻介导的电子传递过程中的作用(图4b):在O₂•⁻与Q•⁻共同存在的条件下,添加H₂可显著提高Q向QH₂的转化率(约翻倍),这表明Q•⁻作为介质在H₂驱动的氧化还原循环中发挥关键作用。
综上,研究结果支持一个连贯的机制模型:在该模型中,Q•⁻作为多功能、无催化金属的中间体,可实现H₂的活化,并根据氧化还原环境的不同,介导不同的电子传递途径。
讨论
总体意义与局限性
本研究的所有实验均在无膜、溶液相的体外体系(XO/Hx体系和KO₂体系)中进行。因此,本文提出的机制解释应被视为可验证的假说,而非已确立的生物学事实。数据显示,在体外条件下,H₂可调控O₂•⁻的动力学,并促进Q还原为QH₂,这一结论得到了曲线下面积(AUC)/初始速率统计分析以及ESR、HPLC交叉验证结果的支持。这些发现证实了溶液体系中H₂驱动Q氧化还原循环的化学可行性;但其生理相关性仍需在线粒体和细胞中进一步验证。因此,线粒体复合体I的示意图仅作为概念图包含在补充材料中(补充图S1)。
生物学相关性与未来方向
尽管溶液相模型证实了H₂参与Q氧化还原循环的可行性,但其生理相关性尚未得到证实。现有数据与“Q•⁻介导的隧穿辅助途径”相符,但无法直接证明该途径的存在。在此前提下,本研究提供了一个溶液相实验框架和可验证的、具有假说生成意义的证据,可为未来研究提供指导,助力开展针对性的体内/原位验证实验,以阐明H₂调控氧化还原平衡的生物学机制。
有趣的是,近期研究表明,Q本身的生物学效应也并非总是呈现简单的剂量依赖性益处。例如,在模式生物中部分抑制Q的生物合成,反而可能延长寿命,这可能是由于活性氧(ROS)水平适度升高并发挥信号分子作用所致(Wang等,2024)。本研究观察到的“H₂对ROS的双相效应(依赖于Q的可得性)”与这一观点一致,强调了精确氧化还原平衡的重要性,而非简单的剂量-反应关系。
未来研究应采用具有呼吸活性的分离线粒体以及纯化的线粒体呼吸链复合体I和复合体III,直接评估不同H₂浓度如何影响外源性和内源性Q/QH₂比值以及线粒体ROS(尤其是O₂•⁻)的生成,特别是在生理和病理相关条件下(如缺氧、假性缺氧或琥珀酸诱导的反向电子传递(RET))。此类实验目前已在本实验室开展,将为H₂介导的隧穿氧化还原调控提供进一步验证,并拓展其生物学意义。
除生物学意义外,无金属参与的H₂活化途径的发现还为绿色化学和可持续能源技术提供了重要潜力。工业上H₂的活化通常依赖稀有金属催化剂(如铂、镍等)实现H₂的裂解。本研究结果表明,在温和条件下,半醌自由基(Q•⁻)或超氧阴离子(O₂•⁻)等有机自由基中间体可能为无金属H₂活化提供替代途径。尽管Q•⁻等瞬时自由基因其不稳定性,可能无法直接应用于工业生产,但这一概念可为开发稳定的“基于有机自由基的仿生催化剂”或“ redox活性聚合物”提供启发,这类材料或可介导隧穿电子传递。此外,类似于Q/QH₂对的两步电子传递体系,可能绕过H₂直接氧化的高活化能垒,为电化学能量转换和储氢技术的发展提供新方向。
综上,这些结果不仅从根本上深化了我们对线粒体氧化还原生物学及相关生物学现象的理解,还为针对“氧化应激相关线粒体功能障碍”的新型治疗策略以及可持续技术创新提供了潜在可能。
与卟啉/血红素介导机制的关系
本研究的无膜溶液体系不含血红素蛋白,因此观察到的钟形/U形动力学行为无需卟啉辅因子参与,且与半醌介导的电子传递一致。尽管如此,在生物学环境中,卟啉/血红素中心介导的途径(包括缺氧条件下铁卟啉对H₂的氧化还原活性以及CO₂向CO的转化(Jin等,2023))仍可能存在,且可能与线粒体中的醌途径相关。
特别值得注意的是,本研究关于Q/Q•⁻/QH₂化学性质的发现,让人联想到复合体III中的Q循环——如前文所述(Ishibashi,2019),在该循环中,细胞色素bL与半醌之间的氧化还原反应发挥关键作用。若聚焦于复合体III中Q•⁻介导的H₂活化,研究Q•⁻与细胞色素bL中血红素之间的电子传递,将为理解H₂在复合体III中调控氧化还原平衡的机制提供重要启示,但这超出了本溶液相研究的范围。
研究局限性
1. 分子氢(H₂)浓度范围
本研究检测了4个离散浓度的H₂(0、0.2、0.4、0.8 mM)的效应,明确观察到了钟形和U形动力学曲线。然而,在关键转变点附近采用更精细的浓度梯度,将能更精确地确定最优H₂浓度以及马库斯正常区与反转区之间的准确阈值。未来研究可采用更小的浓度增量,并将浓度范围扩展至1 mM以上,以明确这些关键点及其在不同氧化还原条件下的变化。
2. 潜在的酶或金属干扰
本研究采用的酶促体系(Hx/XO)包含黄嘌呤氧化酶(XO),该酶含有钼辅因子和铁硫簇。尽管在有氧条件下,XO主要通过底物(Hx)氧化生成O₂•⁻,但无法完全排除H₂与这些金属辅因子发生非预期相互作用的可能性。尽管可能性较低,但如果XO或痕量金属污染物表现出类氢化酶活性,可能会对结果产生影响。不过,在严格无酶的化学体系(KO₂体系)中,我们也观察到了类似的马库斯型动力学和QH₂生成,这有力地表明所提出的H₂活化机制本质上是无金属依赖的。
3. 半醌自由基(Q•⁻)的瞬时检测
由于Q•⁻在水溶液中的寿命极短,且与O₂•⁻共存,直接通过实验检测水溶液中的Q•⁻面临较大挑战。因此,我们通过基于ESR的DPPH自由基清除实验以及O₂•⁻生成调控,间接推断Q•⁻的存在。尽管这些间接方法为结论提供了一致的证据,但直接光谱检测将进一步强化机制解释的可靠性。未来研究可采用先进的高时间分辨率技术(如脉冲辐射分解或快速冷冻淬灭电子顺磁共振(EPR)),直接监测瞬时Q•⁻的生成,并明确H₂活化过程中涉及的精确反应中间体。
4. 泛醇(QH₂)检测的变异性
观察到的QH₂生成量存在显著变异性(对照组变异系数高达100%),这反映了无结构组织的非酶促实验体系中,隧穿介导电子传递过程的固有随机性。这种变异性使得对“H₂驱动Q还原为QH₂的效率”进行定量解释变得复杂。未来研究可采用温度依赖性分析或氘代氢(D₂)的动力学同位素效应实验,进一步阐明这种变异性。若能观察到同位素效应减弱或温度独立性,将为这些反应中隧穿机制的存在提供更明确的证据。
结论
综上,在无膜溶液体系中,H₂可调控Q•⁻的动力学,并在无催化金属或氢化酶的条件下促进Q还原为QH₂。观察到的钟形和U形曲线随Q和H₂浓度的变化趋势,与马库斯电子传递理论框架一致,且符合Q•⁻参与的隧穿辅助途径。这些数据证实了体外溶液体系中H₂驱动Q氧化还原循环的化学可行性;其体内相关性仍有待验证,这也为在呼吸线粒体和细胞模型中开展验证实验提供了动力。
数据可用性声明
本研究中提出的原始数据已包含在文章及补充材料中,如需进一步查询,可联系通讯作者。
https://wap.sciencenet.cn/blog-41174-1509046.html
上一篇:首个慢性疲劳综合征血液检测方法问世
下一篇:人工智能能否真正拥有创造力?