博文
宏基因组常用分析流程简介
|
比对鉴定,拼接组装,功能分析
我们简要介绍两个常用的宏基因组数据分析工具包。
bioBakery workflows
bioBakery来自于The Huttenhower Lab,开发者们提供了多种多样的宏基因组数据分析工具,特别地,搭建了一套workflow以用于处理16S、宏基因组、宏转录组等相关数据,完成序列质控、宿主去除、谱系鉴定、丰度分析、功能预测等工作。整体流程主要基于reads mapping的策略,但也可以通过添加参数--run-assembly完成基因组组装。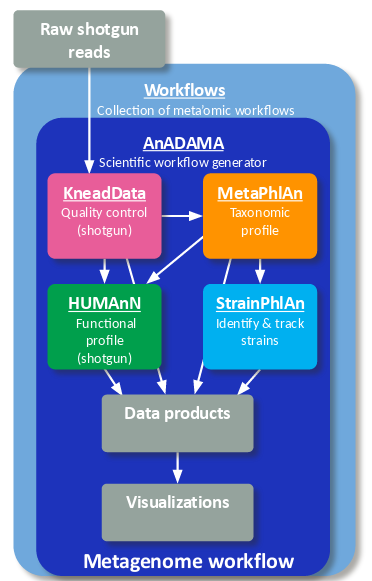 bioBakery workflows可以通过conda、pip或docker一键式安装,用下述命令
bioBakery workflows可以通过conda、pip或docker一键式安装,用下述命令
biobakery_workflows wmgx --input $INPUT --output $OUTPUT --run-assembly
一键式运行,还可根据数据处理结果一键式生成pdf或html报告,工业化标准,简直棒棒哒。相应数据库的下载可单另进行,在docker image中,新建如下文件夹,并设置数据库路径:
mkdir opt/biobakery_workflows_databasesmkdir opt/biobakery_workflows_databases/humann2mkdir opt/biobakery_workflows_databases/humann2/utility_mappingmkdir opt/biobakery_workflows_databases/humann2/chocophlanmkdir opt/biobakery_workflows_databases/humann2/unirefmkdir opt/biobakery_workflows_databases/kneaddata_db_human_genomemkdir opt/biobakery_workflows_databases/strainphlan_db_markersmkdir opt/biobakery_workflows_databases/strainphlan_db_referencetar -xzf full_chocophlan_plus_viral.v0.1.1.tar.gz -C /opt/biobakery_workflows_databases/humann2/chocophlantar -xzf uniref90_annotated_1_1.tar.gz -C /opt/biobakery_workflows_databases/humann2/unireftar -xzf full_mapping_1_1.tar.gz -C /opt/biobakery_workflows_databases/humann2/utility_mappingtar -xzf Homo_sapiens_hg37_and_human_contamination_Bowtie2_v0.1.tar.gz -C /opt/biobakery_workflows_databases/kneaddata_db_human_genome bowtie2-inspect /opt/conda/bin/metaphlan_databases/mpa_v20_m200 > /opt/biobakery_workflows_databases/strainphlan_db_markers/all_markers.fasta humann2_config --update database_folders utility_mapping /opt/biobakery_workflows_databases/humann2/utility_mapping humann2_config --update database_folders protein opt/biobakery_workflows_databases/humann2/uniref humann2_config --update database_folders nucleotide /opt/biobakery_workflows_databases/humann2/chocophlan
MetaWRAP
MetaWRAP整合了基于assembly策略的常用软件工具,让宏基因组数据从质控、到组装、到分类、到功能分析的各个部分,实现一键式、模块化运行。MetaWRAP可以通过conda或者docker安装,数据库需单另下载,并手动修改config-metawrap文件。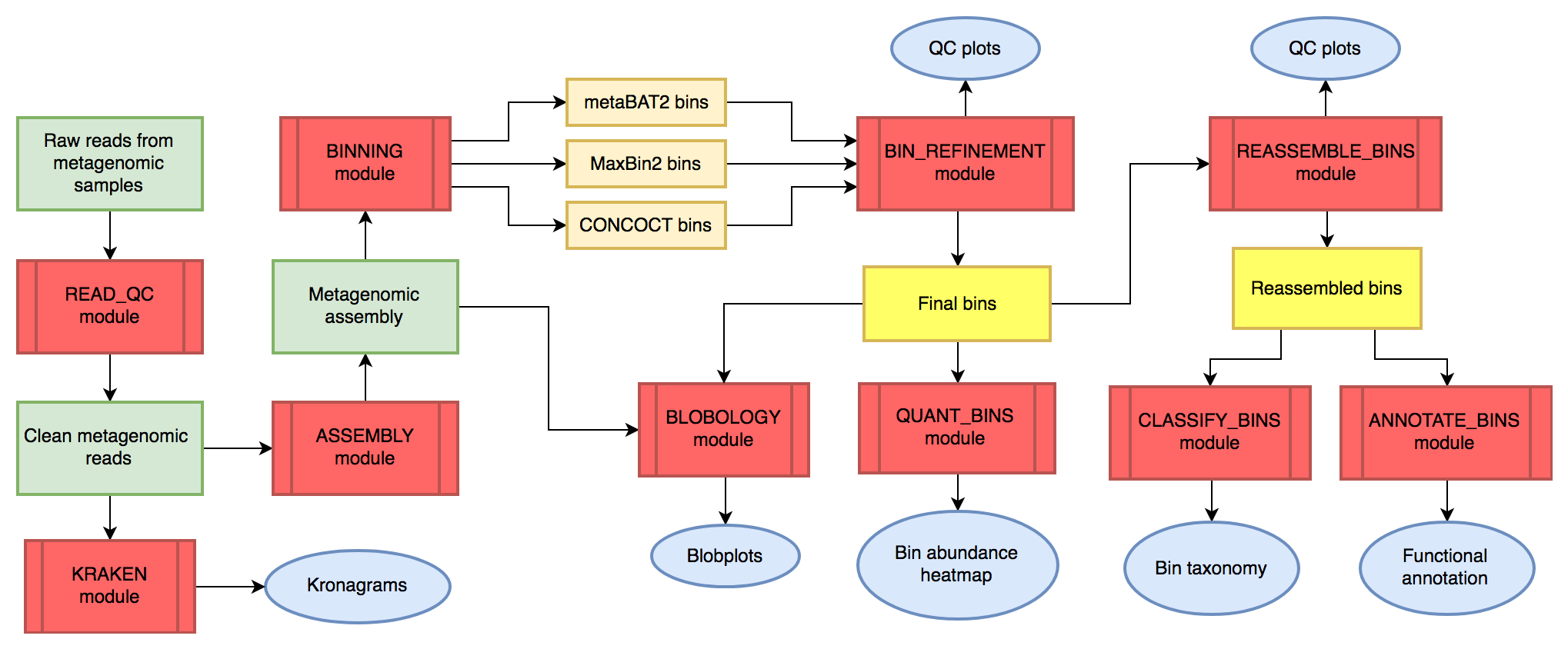
| read_qc | 序列质控、接头去除、宿主去除 |
| assembly | 利用MegaHit或metaSPAdes进行序列拼接 |
| kraken | 利用kraken分析reads、contigs的物种来源、比例 |
| binning | 利用CONCOCT、MaxBin、metaBAT进行binning |
| bin_refinement | 从上一步中整合出“更好的”binning结果 |
| blobology | 可视化bins和物种分类的信息,考察binning的效果 |
| quant_bins | 考察bins在不同样本间的丰度情况 |
| reassemble_bins | 利用bin_refinement整合后的bins进一步组装 |
| classify_bins | 搜索NCBI的nt、tax库进行分类分析 |
| annotate_bins | 功能注释 |
要注意的是,MetaWRAP本身存在着些许bug,比如其目前只接受paired-end测序数据,但其中一些软件实际也可接受single-end,这时就需要手动coding了。同时,各工具也在不断升级换代,很多命令或参数可能过时,比如Kraken已经有2.0版本了,可以选择重新安装或coding。如果是docker的话,MetaWRAP的相关脚本位于/usr/local/bin/下的metawrap-modules和metawrap-scripts文件夹。尽管存在缺点,MetaWRAP依然不失为一款很好的工具流程包,可以为我们的分析带来极大的参考和帮助。
原文链接https://wenlongshen.github.io/2020/03/03/MetaGenomics-Workflows/
https://wap.sciencenet.cn/blog-543513-1228321.html
上一篇:宏基因组分析方法初探
下一篇:关于人类基因组的一些说明