博文
生物信息分析流程(2) WDL入门
|
The Workflow Description Language (WDL) makes it straightforward to define analysis tasks, chain them together in workflows, and parallelize their execution.
对于不同性质的数据,我们面临着不同流程、不同工具、不同参数的选择,一套合适的流程化数据处理框架至关重要。Broad Institute可谓业内之翘楚,我们利用其开发的WDL和Cromwell来搭建自己的生物信息分析流程。本文只做简单介绍,详细文档还是参考官网吧。
WDL基本结构
可以遵循编程语言的基本思想来理解WDL,其脚本主要有5个核心组件,顶层组件:workflow,task,call,任务级组件:command,output。注意并没有显式命名的input,而是通过参数形式传入。其中workflow定义了整个工作流程,类似于main;task定义了单独的每个子任务,位于workflow模块外部,类似于函数;call位于workflow模块内部,表示执行一个特定的函数(task)。注意WDL不是顺序执行的,所以workflow、task、call在脚本内的排列顺序并不重要。图例如下: command和output主要位于task模块内,一个指明了要进行哪些命令操作,另一个则标识出输出文件(为了跟下一个任务串联)。图例如下:
command和output主要位于task模块内,一个指明了要进行哪些命令操作,另一个则标识出输出文件(为了跟下一个任务串联)。图例如下: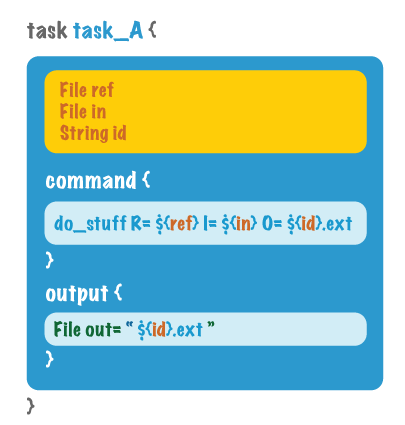
变量设置
先说类型,常用的基本类型有字符串String,浮点数Float,整型Int,布尔Boolean,文件File;数据结构有数组Array,字典Map,对象Object。
变量声明的方法为:Type variableName,实际应用中我们往往不希望每次都设置一遍变量的输入值,而是赋予一些默认值,这时可使用Type? variableName,在随后的命令中通过${default="value" variableName}设置即可。
任务串联
这个是WDL重要的特性之一,可以有多种方式让我们的流程充分运转。
线性:
call stepB { input: in=stepA.out }
call stepC { input: in=stepB.out }多个输入/输出:
call stepC { input: in1=stepB.out1, in2=stepB.out2 }分支:
call stepB { input: in=stepA.out }
call stepC { input: in=stepA.out }
call stepD { input: in1=stepC.out, in2=stepB.out }迭代整合:
Array[File] inputFiles
scatter (oneFile in inputFiles) {
call stepA { input: in=oneFile }
}
call stepB { input: files=stepA.out }别名:
call stepA as firstSample { input: in=firstInput }
call stepA as secondSample { input: in=secondInput }
call stepB { input: in=firstSample.out }
call stepC { input: in=secondSample.out }灵活选择,灵活运用,让流程跑起来!
语法检查
官方贴心地提供了wdltool,通过java -jar wdltool.jar validate myWorkflow.wdl来检查语法。
参数设置
各种参数和输入都可以在WDL脚本内设定,但为了可读性和易用性,我们建议使用JSON文件来指定各个参数。同样地,可利用wdltool,通过java -jar wdltool.jar inputs myWorkflow.wdl > myWorkflow_inputs.json生成一个JSON文件作为模板,后续更改相应参数即可。
流程执行
通过Cromwell启动整体流程,一个基本的命令是:java -jar Cromwell.jar run myWorkflow.wdl --inputs myWorkflow_inputs.json,终端会输出相关信息,但所有的输入文件、输出文件和日志都会保存在特定文件夹内。
实例
我们接上一篇中用到的两个docker,构建一个简单的ChIP-seq数据分析流程(注意这里只做了mapping和peak calling,并不是完整的流程)。我们把WDL相关工具放在/home/wenlong/WDL/下,数据放在/home/wenlong/Data/下。注意目前WDL相关功能还在完善,比如生成hash code会在指定bowtie2的index时造成一些麻烦(bowtie2 -x参数是index名字,而非实际文件,而WDL使用docker时,由于有hash code而难以指定index名字,故我暂时调整docker,在原工作目录/data下加入hg19的bowtie2 index。后续再研究研究别的方法。同时要注意现在WDL需要docker image有sha256)。构建WDL脚本ChIPseq_demo.wdl如下:
## Copyright Wenlong Shen, 2018
##
## Cromwell version support
## - Successfully tested on v35
# WORKFLOW DEFINITION
workflow ChIPseq_demo {
File sample_1_r1
File sample_1_r2
String sample_name
String? sample_format
String index
String macs2_output_dir
String bowtie2_docker
String macs2_docker
Int cpu_num
# Map reads to reference
call Mapping_Bowtie2 {
input:
sample_1_r1 = sample_1_r1,
sample_1_r2 = sample_1_r2,
sample_name = sample_name,
index = index,
docker_image = bowtie2_docker,
cpu_num = cpu_num
}
# Call peaks
call PeakCalling_Macs2 {
input:
sample_sam = Mapping_Bowtie2.output_sam,
sample_format = sample_format,
sample_name = sample_name,
macs2_output_dir = macs2_output_dir,
docker_image = macs2_docker,
cpu_num = cpu_num
}
}
# TASK DEFINITIONS
# Run Bowtie2
task Mapping_Bowtie2 {
File sample_1_r1
File sample_1_r2
String sample_name
String index
String docker_image
Int cpu_num
command {
bowtie2 -x ${index} -1 ${sample_1_r1} -2 ${sample_1_r2} -S ${sample_name}.sam -p ${cpu_num}
}
runtime {
docker: docker_image
}
output {
File output_sam = "${sample_name}.sam"
}
}
# Run Macs2
task PeakCalling_Macs2 {
File sample_sam
String? sample_format
String sample_name
String macs2_output_dir
String docker_image
Int cpu_num
command {
macs2 callpeak -t ${sample_sam} -f ${default="SAM" sample_format} -n ${sample_name} --outdir ${macs2_output_dir}
}
runtime {
docker: docker_image
}
output {
File output_narrowPeak = "${macs2_output_dir}/${sample_name}_peaks.narrowPeak"
}
}相应的json文件ChIPseq_demo.inputs.json如下:
{
"ChIPseq_demo.sample_name": "test",
"ChIPseq_demo.sample_1_r1": "sample/test.r1.gz",
"ChIPseq_demo.sample_1_r2": "sample/test.r2.gz",
"ChIPseq_demo.index": "/data/hg19",
"ChIPseq_demo.macs2_output_dir": "macs2_result",
"ChIPseq_demo.bowtie2_docker": "wenlongshen/bowtie2.hg19:ubuntu.v1",
"ChIPseq_demo.macs2_docker": "wenlongshen/macs2:ubuntu.v1",
"ChIPseq_demo.cpu_num": 8
}最后运行命令:
java -jar cromwell-35.jar run ChIPseq_demo.wdl --inputs ChIPseq_demo.inputs.json
终端打印相关信息,最终成功生成peaks文件。
总体来说,WDL结构略复杂,槽点很多,容易出错,调试较麻烦,但依然不失为科学共同体杰出的流程化语言,值得学习、推广应用。
原文链接https://wenlongshen.github.io/2018/09/15/Pipelining-Solution-2/
https://wap.sciencenet.cn/blog-543513-1143438.html
上一篇:生物信息分析流程(1) docker入门
下一篇:Feature Importance