博文
[转载]最新研究《Cell》纵向多组学揭示肠易激综合征(IBS)的潜在机制
|| |
谷禾健康
肠道微生物组与多种人类慢性胃肠道(GI)疾病有关。肠易激综合症(IBS)是一种普遍存在的疾病,其特征是反复出现腹痛或不适。IBS主要见于女性,并与粪便形式或频率变化有关,并基于主要便秘形式(便秘为主(IBS-C),腹泻为主(IBS-D)或混合型(IBS-M))。 由于动物和人类研究之间明显的脱节以及缺乏针对疾病特异性生理变化的综合多组学观点,因此很难确定肠道菌群在疾病中的作用机理。

近日,美国梅奥诊所消化内科和肝病科 Purna C. Kashyap研究团队和明尼苏达大学生物科学学院 Dan Knights团队合作在 Cell上发表了题为 Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome 的文章,研究团队对IBS宿主生理进行了多组学测量的纵向研究 (gun弹枪浅宏基因组测序、16S rRNA基因测序、代谢组学研究,细胞因子测量,转录组和甲基化组分析)最终确定了IBS亚类型特异性、症状相关的微生物组成和功能变异,其中一组已确定的微生物代谢产物变异子集与IBS有关的宿主生理机制相关。 通过Lasso回归机器学习方法整合多个数据层,鉴定出的微生物代谢物变化的子集对应于与IBS相关的宿主生理机制。研究团队将嘌呤代谢确定为一种新型IBS宿主-微生物代谢途径,同时嘌呤饥饿被确认为潜在的IBS治疗靶标。
点评总结
这项研究主要采用了纵向多时间点采样,针对IBS这类症状波动较为明显的疾病研究纵向多时间点采样可以减少横断面研究的采样偏差,减少入组病人的数量但提高统计检验的效力。可以看到研究纳入的病人并不很多,但采用平均值之后统计效果明显。
另外研究同时进行了多组学的检测,包括代谢组和黏膜转录组等,通过对宏基因组功能差异的分析以及代谢组的差异分析同时锁定了多项代谢异常,尤其是次黄嘌呤的水平异常。进一步对次黄嘌呤水平与宏基因组的菌种进行关联分析锁定了重要的相关菌,通过SV关联分析,在更细尺度上筛选出了可能的功能基因区段。
这种组合方式提供了完整的研究思路,从人群尺度寻找疾病可能的代谢和生理机制,并经由多组学的关联分析锁定可能的菌种标志,再到精细化筛选出可能的功能基因。
在菌群方面研究采用了宏基因组和16S,对粪便样本采用宏基因组,对黏膜样本采用16S,因为黏膜样本含有较高的人体DNA,16S更为合适。粪便样本的宏基因组直接采用和RefSeq89版本进行比对注释,基因部分同时结合了序列比对和利用基因组数据直接提取注释相结合。宏基因组测序能够提供菌株层面的分辨率,同时也是后续结构变异关联分析的必要条件,随着参考基因集的完善,中等测序深度的浅宏基因组将可以大量应用于这类研究中(浅宏基因组文末详细介绍和福利活动 )。
全文介绍
全文缩略词整理:
肠易激综合征(IBS)
便秘为主的肠易激综合征(IBS-C)
腹泻为主的肠易激综合征(IBS-D)
混合型为主的肠易激综合征(IBS-M)
健康对照(HCs)
症状严重程度评分(SSS)
短链脂肪酸(SCFA)
靶向液相色谱-质谱(LC-MS)
5-羟色胺受体4(5-HT4R)
胆汁酸(BAs)
胆酸(CA)
鹅去氧胆酸(CDCA)
核磁共振氢谱(1H-NMR)
黄嘌呤磷酸核糖基转移酶(XPRT)
嘌呤核苷磷酸化酶(PNP)
缺失区(DR)
可变区(VR)
Bray-Curtis差异(BCD)
不规则性(BCDI)
差异甲基化区域(DMR)
本文用到的实验方法汇总:
样本人群和数据生成
18-65岁的IBS-C和IBS-D患者经过严格的筛查确诊患者入组。IBS-C和IBS-D受试者均符合罗马III级标准。有腹部手术史(阑尾切除和胆囊切除除外)、被诊断为炎症性肠道疾病、显微镜下结肠炎、腹腔疾病或其他炎症性疾病、在过去4周内使用抗生素、出血风险或服用增加出血风险的药物、过去一周准备接受结肠镜检查、怀孕、计划在研究期间怀孕、是易受感染的成年人以及年龄在18岁以下或65岁以上的志愿者被排除在外。
食物频率问卷(FFQ)和24小时饮食回想问卷
动物实验
小鼠实验
Ussing chamber 试验
Ussing Chamber,尤斯灌流室,离是研究跨上皮转运的工具,可用于包括离子转运、营养物质转运及药物转运等的研究。通过跨上皮转运的研究,可以了解上皮的离子通道机制、营养成分及药物透过上皮的吸收、影响上皮屏障功能以及通透性的因素等。本文的Ussing Chamber实验研究了5-羟色胺(5-HT)对结肠上皮短路电流(ISC;一种反映肠道分泌物的跨上皮离子流量的测量)的变化。
微生物组测序
QIAGEN PowerSoil 提取粪便和组织DNA
浅宏基因组使用HiSeq 2500(快速模式)单端读数为100 bp(1x100)和NextSeq 150 bp单端读数(1x150)进行测序。
扩增子测序,对核糖体RNA基因的V4区进行测序,扩增子序列与来自同一细菌基因组的16S rRNA基因在shotgun测序法中使用BURST。
微生物组数据分析
宏基因组部分使用的是浅宏基因组,使用1x100或1x150单端,使用BURST以97%相似度和RefSeq(v86版本)进行比对,过滤比对测序深度低于1万reads的样本。KEGG正射图也可以从参考基因组中进行预测,并利用SHOGUN对预测的图谱进行扩充,以改进对低丰度基因的估计
基因丰度部分是直接比对从refseq基因组中提取的KEGG注释序列,并利用SHOGUN改进低丰度基因的预测)
在测试亚组之间的分类单元差异之前,删除了90%的受试者中不存在的分类单元。 为了识别差异丰富的特征,使用FDR截止值<0.25。
通过提取所有健康对照(HC)与HC或IBS样本之间的成对差异来计算基于Bray-Curtis差异(BCD)的不规则性(BCDI),并存储这些差异的中位数。HC值的第90个百分位数用作鉴定与HC样品不同的微生物组样品的临界值。通过随机抽取每个HC对象一个样本并在这些样本中识别BCDI 500次,对第90个百分位数的临界值进行了敏感性分析。 此外,还计算了平均HC微生物组丰度的第90个百分位数临界值。 使用平均值不会改变第90个百分位数的临界值(0.63)。
代谢组学
血清和粪便样品的核磁共振代谢(1H-NMR)谱分析,测定粪便样品中丙酸、丁酸和醋酸的相对丰度,非靶向1H-NMR谱判别分析(PLS-DA)模型确定了IBS亚组和HC粪便样本之间的代谢变异
通过LC-MS /MS进行胆汁酸分析
使用ACQUITY超高效液相色谱(UPLC)系统和Xevo G2-S四极飞行时间(Q-TOF)质谱仪上对样品进行了分析。
通过GC-MS / MS进行SCFA定量
使用7000D 三重四极杆 GC/MS((Agilent Technologies Ltd.)对SCFA进行定量分析。7000D 三重四极杆气质联用系统是 GC/MS/MS 发展史上迄今为止最为成功的最新型号。使用真正的SCFA标准品绘制了11点校准曲线和合并的QC样品。
通过LC-MS / MS进行色氨酸定量
色氨酸定量使用LC-MS/MS在Waters Acquity UPLC上进行,色谱柱为T3 C18柱(1 ×50 mm,1.7 uM),联用Waters Xevo TQ-S三重四极杆质谱仪。
细胞因子测量
使用多路Luminex定量分析IL-8,IFNγ,IL-10,II-18,IL-22,瘦素,血管内皮生长因子(VEGF),膜结合免疫球蛋白(MIG),IL-1β,IL-17A,IL-1RA,IL-6和 TNFα。使用酶联免疫吸附测定(ELISA)对TGFb-1进行了定量。
RNA测序和分析
提取mRNA, 使用Illumina High Seq-2000测序,使用MAP-RSeq v.2.0.0、edgeR 2.6.2、R包RITAN(https://www.bioconductor.org/packages/release/bioc/html/RITAN.html)进行基因机损,差异表达和路径富集分析。
甲基基因组测序和分析
使用Illumina Infinium甲基化EPIC BeadChip评估基因组DNA中的全基因组甲基化,使用R软件包ChAMP 2.9.10、Combat方法、limma函数和Benjamini-Hochberg(BH)得到甲基化的CpG位点,使用R包RITAN,将与DMC或DMR相关的基因用于途径富集分析。
多组学数据整合
使用R中的Maaslin2软件包(http://huttenhower.sph.harvard.edu/maaslin)研究了粪便微生物特征与粪便代谢产物之间的关联。 使用最小丰度运行Maaslin2,将微生物特征的最小患病率分别设置为0.0001和0.5。 经FDR校正的q值的阈值设置为0.25。 将线性混合效应模型应用于与被设置为随机效应的受试者的关联。
用Lasso回归机器学习方法拟合,鉴定基因-微生物组和基因-代谢物关联,该基因模型使用每个基因的基因表达作为响应,并使用微生物组丰度或代谢物浓度作为预测因子。回归方法是一种对数值型连续随机变量进行预测和建模的监督学习算法,回归分为Linear Regression线性回归,Logistic Regression逻辑回归,Polynomial Regression多项式回归,Stepwise Regression逐步回归,Ridge Regression岭回归,Lasso Regression套索回归,ElasticNet回归。其中,Lasso回归方法是在统计学和机器学习中同时进行特征选择和正则化(数学)的回归分析方法,旨在增强统计模型的预测准确性和可解释性,在实践中,岭回归与套索回归首先岭回归。但是,如果特征特别多,而某些特征更重要,具有选择性,那就选择Lasso可能更好。
体内和体外次黄嘌呤消耗实验
Mega培养基中培养细菌,使用LC-MS测定培养上清液中的次黄嘌呤水平,在6周龄的无菌Swiss Webster小鼠上进行了单菌落实验。
次黄嘌呤(Sigma-Aldrich)管饲喂养后第4天,处死小鼠并收集盲肠内容物,使用Amplex Red黄嘌呤/黄嘌呤氧化酶测定试剂盒(Thermo Fisher)测定盲肠内次黄嘌呤和黄嘌呤的总浓度
结果分析:
一 纵向采样克服了跨部门微生物组研究中的异质性
慢性胃肠道疾病中肠道微生物组的横断面研究提供了高度动态的生态系统的快照。 除了饮食,药物使用,生活方式和其他环境因素的影响外,随着时间的流逝微生物群的变化也可能反映疾病活动的变化。
该文通过对纵向数据进行二次采样,测试显着的分类单元,将单个时间点的结果与受试者平均所有数据所获得的结果进行比较,来评估纵向采样与横截面采样相比对识别成分变化的影响时间点。
比较不同时间点时,在各个时间点观察到的HC和疾病组之间的分类单元丰度差异非常不一致,并且与平均数据中观察到的变化不重叠。 当使用平均数据而不是单时间点数据时,发现多个链球菌属物种的丰度明显更高。与HC相比,IBS-D中新近鉴定出的门合生植物的丰度要低得多。 我们还发现,个体内部差异高于个体内部差异,这支持了我们对每个个体的纵向数据进行平均的方法(STAR方法,t检验p <0.0001)。
这些发现凸显了在慢性疾病中进行纵向采样的必要性,以可靠地识别使用横截面采样可能遗漏的微生物群变化。 因此,我们主要报告时间平均数据的发现。 最近的一项研究进一步证明了这一点,该研究表明,在多个采样时间点使用平均值时,常用的“组学”方法更为准确。
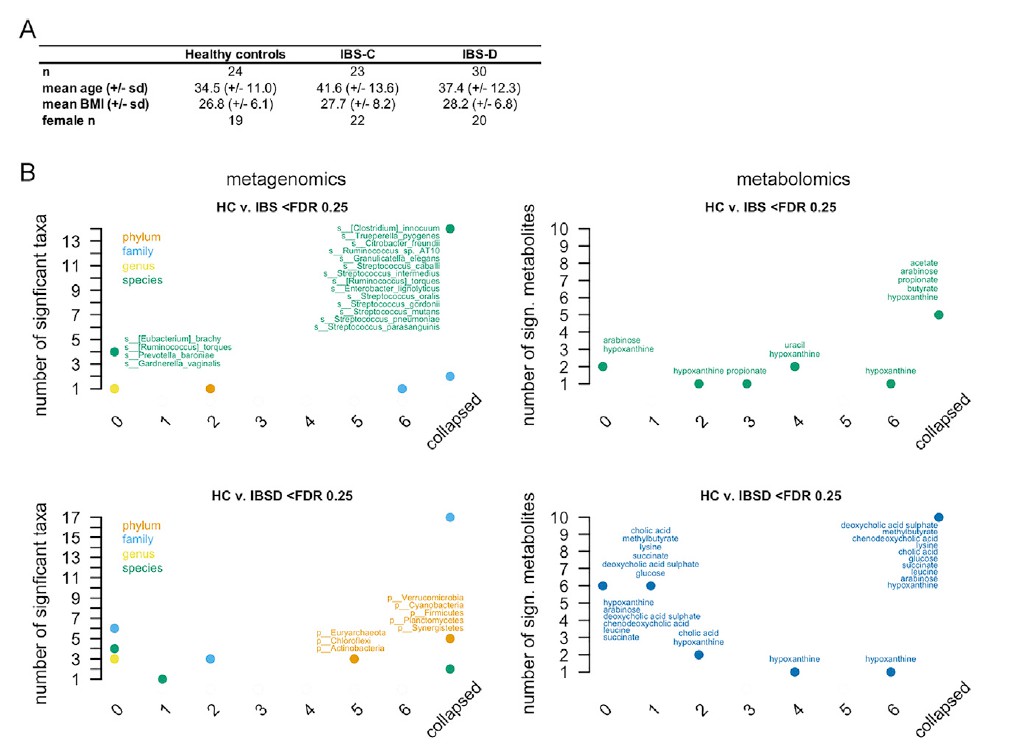

二 纵向采样揭示了随着时间的流逝,IBS-C微生物群具有更大的可变性
与HC和IBS-D受试者相比,IBS-C患者的粪便微生物群组成随时间表现出更大的变异性。 此外,与IBS-D样本相比,平均IBS-C粪便样本的香农多样性更高(ANOVA,Tukey HSD p值为0.016)。
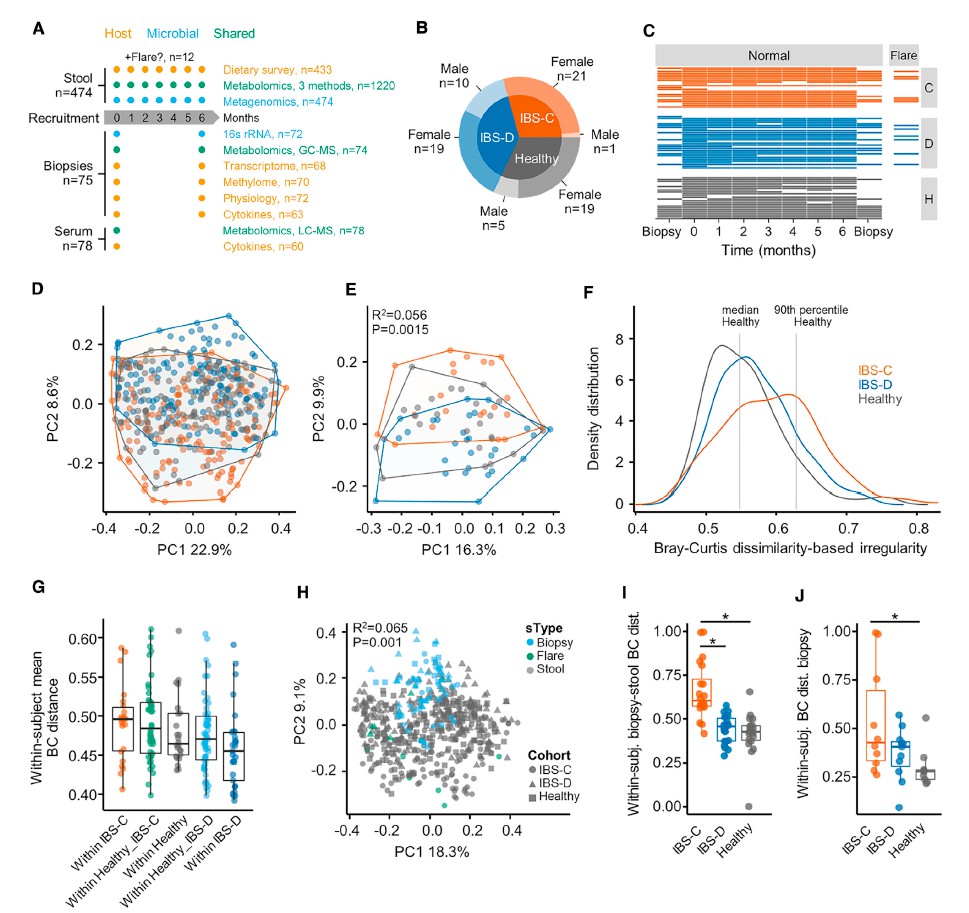
粪便样品中结肠黏膜的微生物组成与腔微生物群有显着差异。 与HC相比,IBS患者的粘膜相关菌群的特征是变形杆菌水平明显较高。 与IBS-D或HC相比,IBS-C患者的与黏膜相关的微生物菌群与其各自的腔内微生物菌群相似性较低。 这可能反映出IBS-C患者菌群的迁移时间较长,而社区之间有更多的时间分化。 此外,IBS-C患者的粘膜相关菌群的个体内变异性随时间变化更大,这与我们在腔菌群中观察到的相似。
三IBS症状严重程度与肠道菌群的功能变化有关
在特定采样点使用IBS SSS(0-500)报告IBS的严重程度,IBS SSS是腹痛强度、频率、肿胀、对排便习惯的不满以及IBS对一般生活的影响的累积度量。我们观察到重度IBS-D(SSS>300)中超过20种乳杆菌的相对丰度高于轻中度IBS-D(SSS<300)。而且这与受试者食用益生菌或乳制品无关。
在粪便宏基因组学功能富集分析时,我们发现在FDR中74个KO与严重的IBS-C相关,而44个与严重的IBS-D相关。 重度IBS-C和IBS-D中均发现了乙醇脱氢酶(ADH)的KO途径,且在重度IBS-C和IBS-D比轻中度IBS中高。 乙醇脱氢酶(ADH)显示与双歧杆菌和链球菌属呈正相关。这些数据表明ADH活性可能与腹痛有关,腹痛是IBS-C和IBS-D共同的主要症状。
四 代谢组学与生理学结果阐明了肠道微生物群代谢对胃肠功能的影响
H1核磁共振(NMR)光谱显示,与HC相比,IBS-C患者粪便样本中的短链脂肪酸(SCFA)丙酸酯,丁酸酯和乙酸酯显着降低。与腔内代谢产物一致,与HC组相比,IBS-C组的结肠黏膜活检样品中的乙酸盐(通过气相色谱-质谱[GC-MS]测定)也显着减少。,SCFA的这些差异与膳食纤维的总摄入量无关,因为这在各组之间没有显着差异。
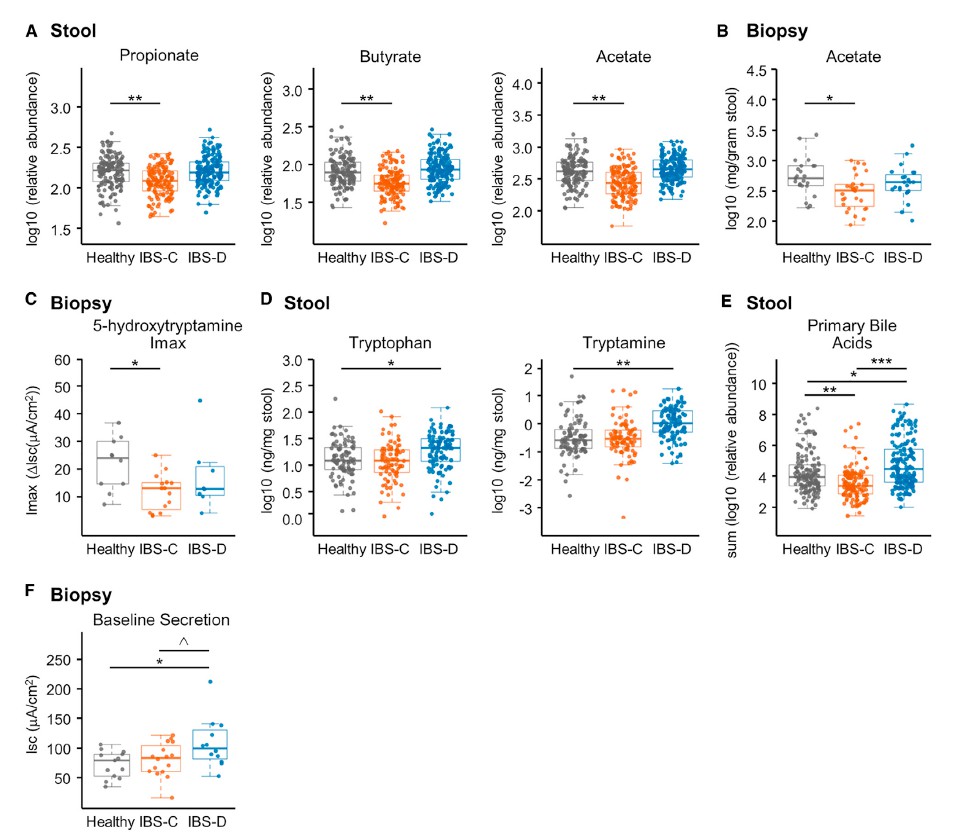
Ussing chamber 试验结果表明水分流伴随离子通量,并且分泌减少会导致便秘中粪便含水量降低。 相反,增加的离子通量可导致分泌更多的水,导致腹泻。色胺均显着增加了结肠分泌且在各组之间没有显着差异,表明,IBS患者和HCs的结肠上皮能够由色胺引起的液体分泌,因此观察到的变化可能是由于色胺的丰度变化导致。

进一步使用靶向液相色谱-质谱(LC-MS)方法研究了粪便样品中色胺和其他色氨酸代谢物的变化。 发现,IBS-D患者粪便样品中的色氨酸和色胺都显着增加,因此可能部分归因于IBS-D粪便中水分的增加。 我们证实这些代谢物的变化与蛋白质摄入的饮食差异无关。
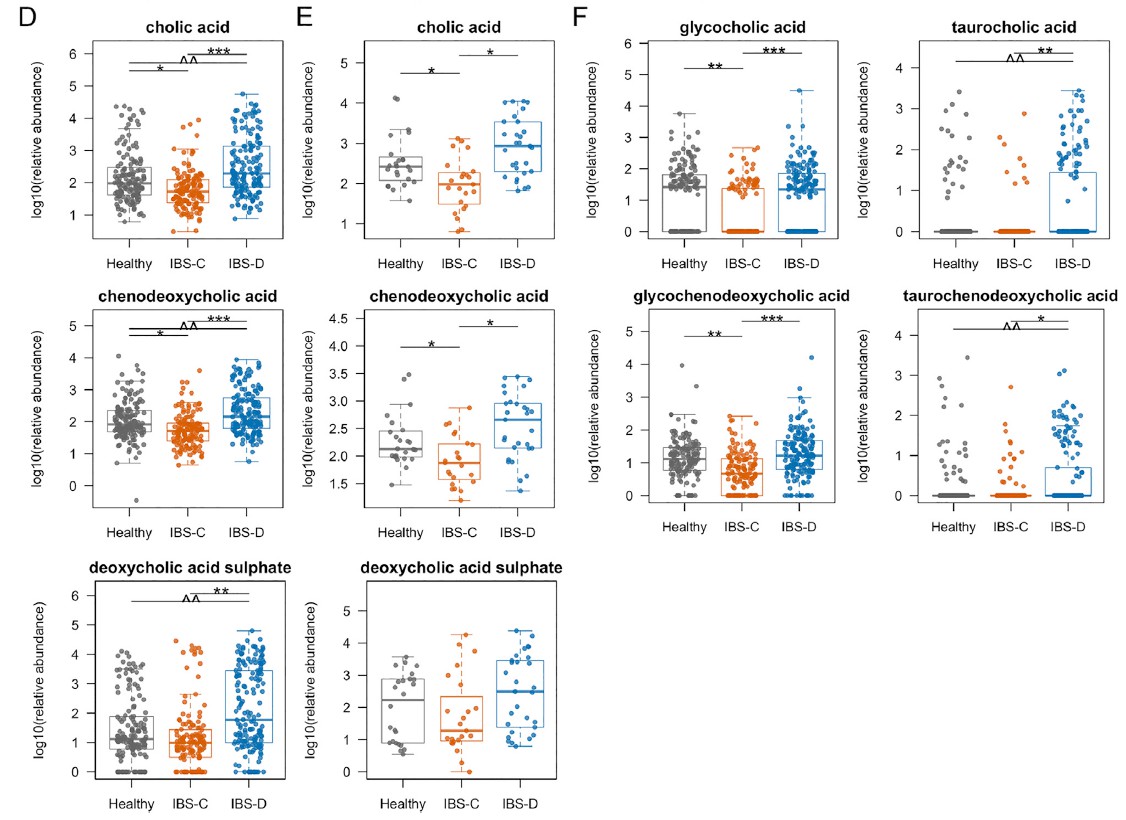
使用LC-MS / MS我们IBS-D患者粪便样品中未结合的初级BA含量明显较高,而IBS-D患者粪便样品中未结合的初级BAs含量明显较低。与HC和IBS-C受试者相比,IBSD中个体初级共轭和非共轭BA和DCA-S的量更高。 由于像CDCA这样的羟基化初级BA可能会增加结肠分泌,因此运用Ussing chamber测试了CDCA在无菌小鼠结肠黏膜下黏膜下层制剂中的作用,结论支持了CDCA水平升高在增加IBS-D患者粪便中水分含量方面的生理作用。
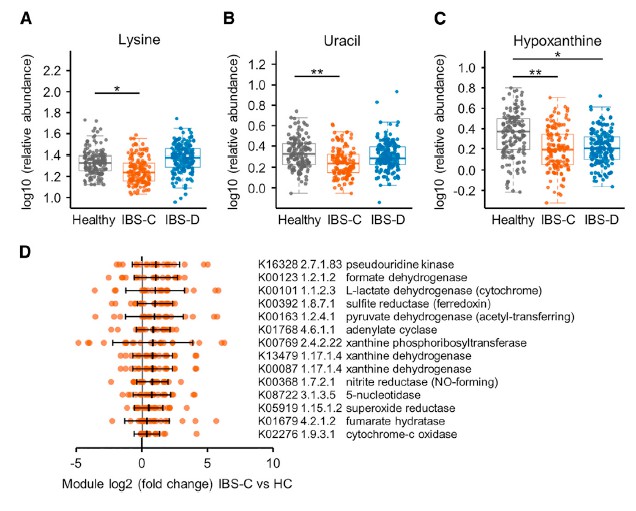
五 联合多组学分析确定了IBS中的新型微生物代谢途径
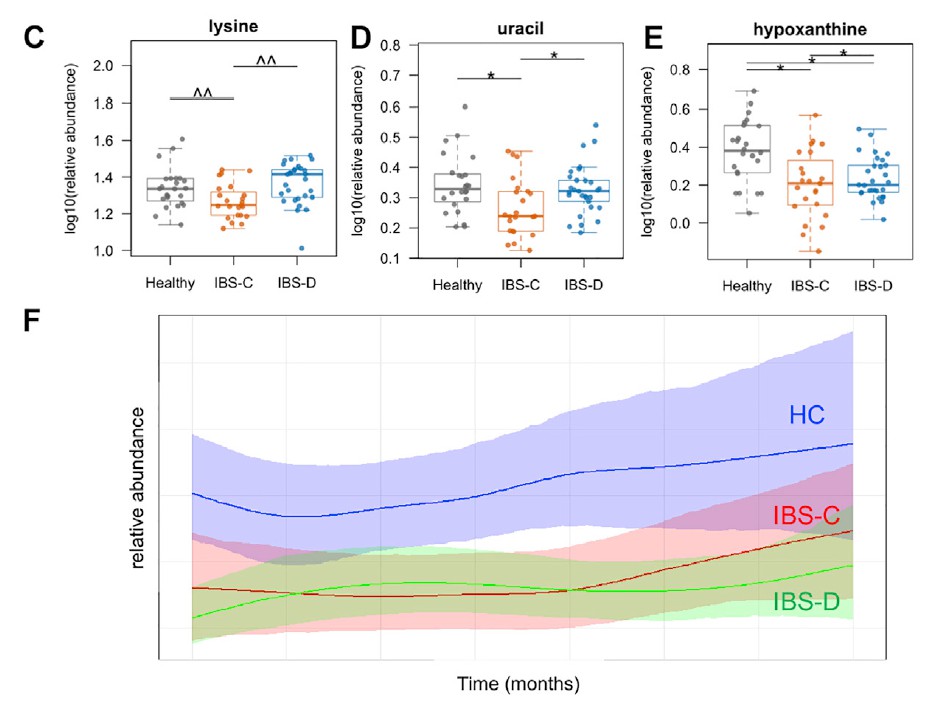
采用了非靶向代谢组学方法来鉴定可能导致IBS病理生理变化的新型微生物途径。基于无目标1H-NMR光谱图的潜在结构判别分析(PLS-DA)模型的投影确定了IBS亚组和HC粪便样品之间的代谢变化。 与HC相比,IBS-C患者粪便样品中的赖氨酸,尿嘧啶和次黄嘌呤均显着降低。 IBS-D患者的次黄嘌呤含量也较低,尽管与IBS-C的意义不同。
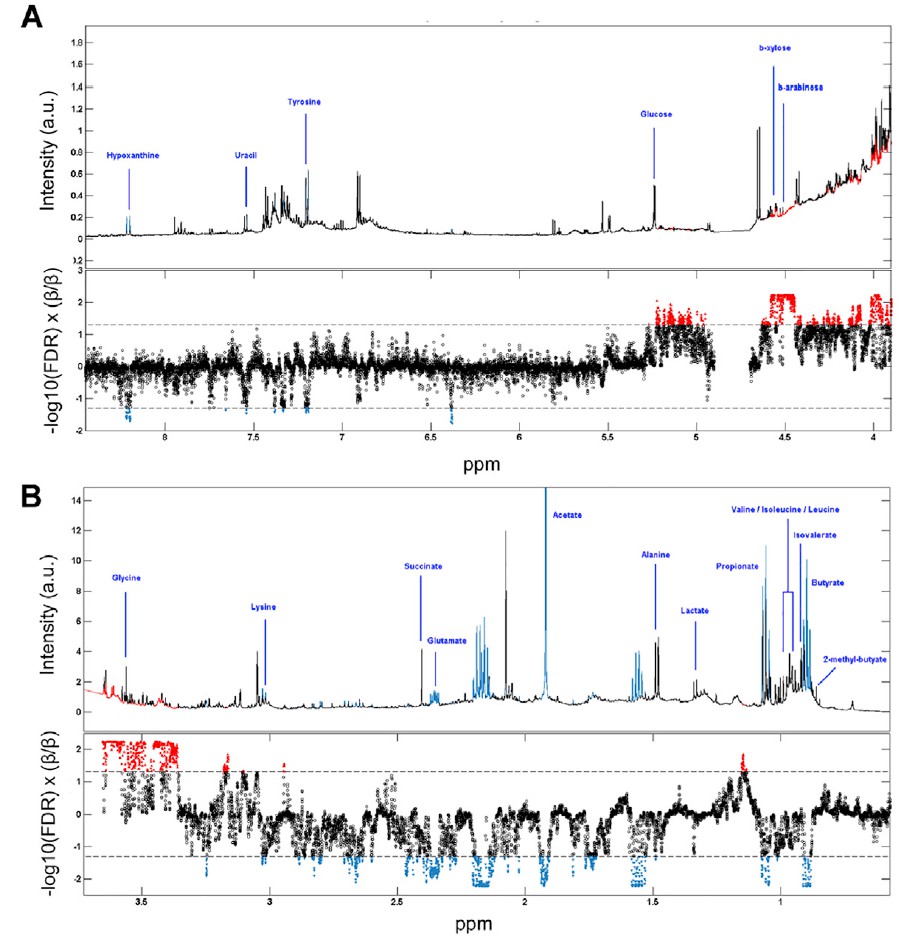
分析IBS和HC患者粪便样本中次黄嘌呤相关的宏基因组学功能,发现IBS-C中的黄嘌呤脱氢酶/氧化酶(XO; 1.17.1.4)和黄嘌呤磷酸核糖基转移酶(XPRT; 2.4.2.22)途径相对于HC有所升高。 XPRT从黄嘌呤单磷酸中释放出黄嘌呤,这是抢救嘌呤的第一步。 在下游,XO是一种具有低底物特异性的酶,可作用于黄嘌呤或次黄嘌呤以产生尿酸。 这些较高水平的XPRT和XO模块表明,IBS患者中肠道菌群对嘌呤的分解作用增加。
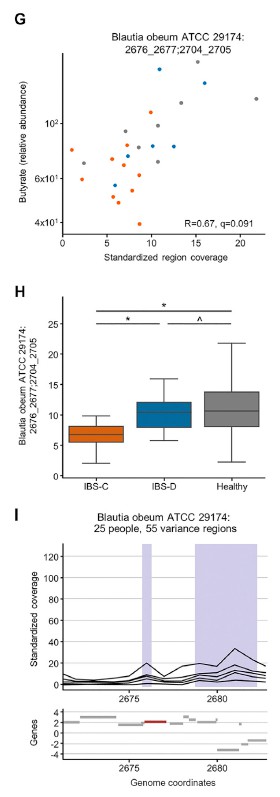
进一步检查了宏基因组的KO分析,以探索次黄嘌呤代谢的两个方面,即其在调节上皮能量状态中的作用以及在假定的氧化剂作用下生成H2O2和超氧阴离子。 与HC相比,IBS-C粪便中C氧化酶的丰度明显更高。 有趣的是,IBS-C中的超氧化物还原酶(1.15.1.2)升高,这可能反映了IBS-C肠道微生物组应对氧化应激的能力增强。 在XO活跃的情况下,这可能是必要的。
综上所述,这表明IBS患者的微生物组对次黄嘌呤的利用和分解能力增强,这与IBS-C粪便中次黄嘌呤水平的降低是一致的。
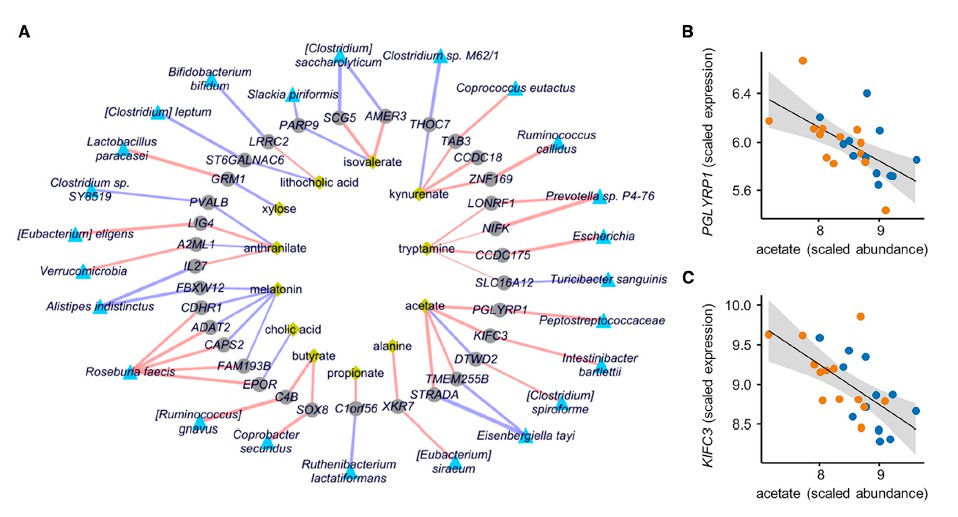
六 胆汁酸,丁酸和次黄嘌呤代谢有关
为了进一步阐明微生物对IBS中鉴定的代谢物丰度的贡献,首先基于线性模型(Maaslin2; http://huttenhower.sph.harvard.edu/maaslin)进行了直接多元相关分析。 这确定了HC样品有60种重要的代谢物种类相关性,IBS-C有28种,IBS-D有46种。 所有组均无相关性。 HC和IBSC或IBS-D中存在12个。 IBS-C和IBS-D子组都存在两种相关性。
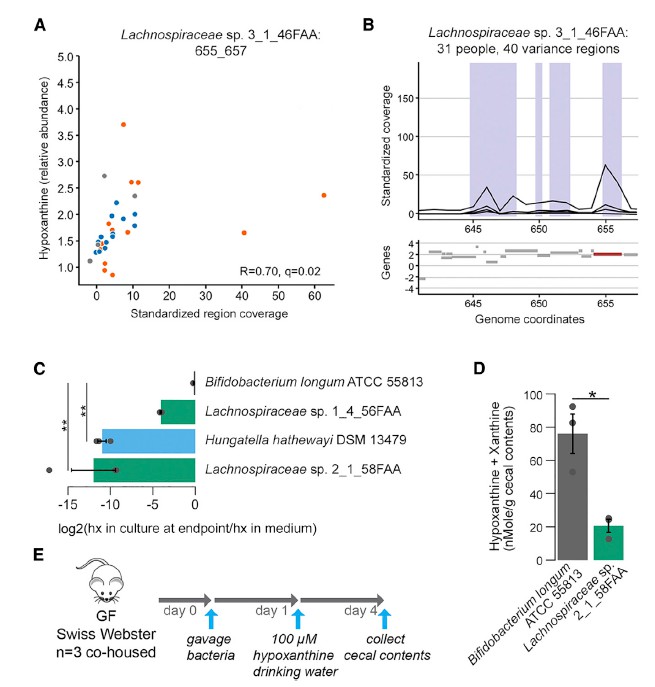
尽管以上相关方法使我们能够识别粪便代谢物差异的潜在微生物驱动因素,但无法识别可能与检测到的代谢物差异相关的特定微生物基因。 因此,我们使用最近描述的将结构可变的基因组区域与代谢物丰度相关联的方法(SV关联),测试了可能导致各组之间代谢输出变化的特定细菌基因组区域。
七 微生物代谢有助于次黄嘌呤水平
为了更深入地了解微生物组在降低次黄嘌呤水平中的作用,选择了与Lachnospiraceae sp的基因组相似性选择了2个Lachnospiraceae菌株进行无菌小鼠实验,结果表明现定植了Lachnospiraceae sp的小鼠的盲肠内次黄嘌呤水平明显降低。 与长双歧杆菌定植的小鼠相比为2_1_58FAA(图4E)。 由于常规使用次黄嘌呤水平会增加,这表明微生物确定的次黄嘌呤水平是微生物生产与消耗之间平衡的结果。
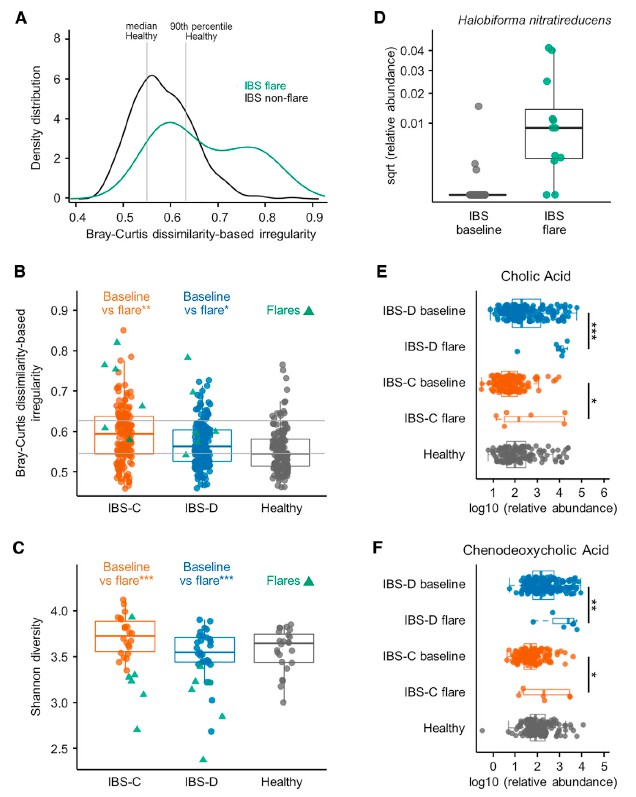
八 IBS患者爆发时肠道微生物组和微生物代谢产物的改变
IBS是一种症状严重程度随时间变化的慢性疾病,大多数患者会出现短暂的症状恶化。前面纵向分析确定了肠微生物组与IBS患者症状严重程度之间的潜在联系。对个别报告中显示恶化时收集的粪便样本进行分析,与非爆发基线组合IBS样本相比,爆发期样本显示出更高的BCDI),与各个IBS子组的平均样本相比,爆发期样本的Shannonα多样性更低(图5C)。 在将IBS患者作为一组时以及在IBS-D和IBS-C患者中,特定细菌类群都与耀斑显着相关(IBS-D和IBS-C患者为168种,IBS-C为40种,IBS-D为7种) 与各自平均基线样本相比,来自Mann-Whitney U检验的q <0.1(表S2)。 这些重要物种在爆发期间几乎普遍减少。发现包括色胺,CA和CDCA在内的分泌代谢物在亚组中升高爆发时IBS患者的比例(BA为6/11,色胺为4/11)。 这些观察结果表明,不同患者的症状恶化可能是独特的微生物和代谢特征。

九 微生物组和代谢组学数据与转录组学和表观遗传学差异的整合揭示了IBS中新型的宿主-微生物组相互作用
对于大多数慢性疾病,IBS的病理生理是多因素的,其来自宿主途径,微生物途径和宿主-微生物共代谢。 为了确定微生物代谢对宿主功能的影响,我们首先比较了在结肠活检组织中观察到的转录和表观遗传学变化。 还通过构建跨组学相关网络,将转录组数据与代谢物和微生物群的丰度相集合,从而以无针对性的方式,使用这些数据来确定推定的宿主-微生物-代谢相互作用。
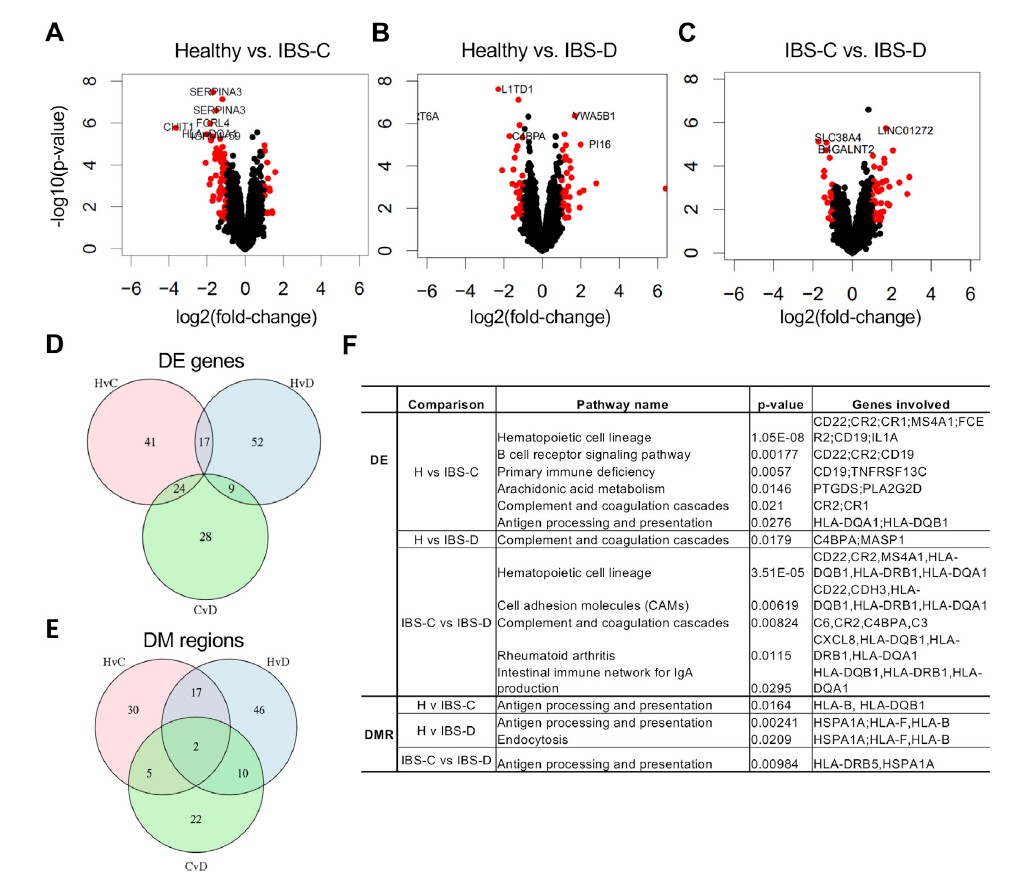
十一 多组学集成确定结肠上皮中的嘌呤饥饿是潜在的新机制IBS
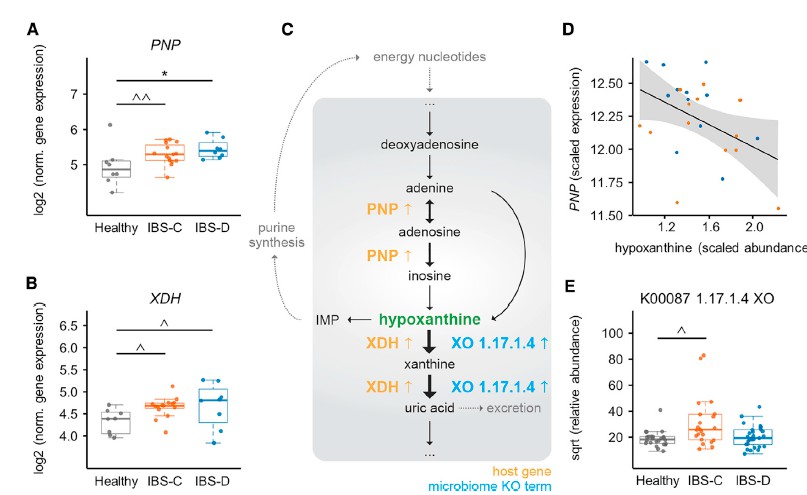
前面确定了IBS-C和IBS-D中粪便次黄嘌呤的含量显着降低,确定了微生物次黄嘌呤的降解导致肠道次黄嘌呤的水平降低,并确定了功能性变化,表明IBS-C粪便中微生物组导致嘌呤降解的增加。 然而,由于次黄嘌呤是宿主-微生物共代谢物,因此其库可同时受到微生物和宿主代谢的影响。
其实,肠上皮细胞的嘌呤从头合成能力有限,而是主要依靠挽救途径进行腺苷酸的生物合成,因此,为了确定由于次黄嘌呤池的消耗而导致宿主的继发效应,需要进一步确认嘌呤挽救途径中可能的转录变化。嘌呤挽救途径中的第一个基因嘌呤核苷磷酸化酶(PNP)在IBS-C和IBS-D中均表达高2倍。在IBS患者中PNP表达呈与次黄嘌呤水平呈负相关。 重要的是,宿主遗传学的变异不负责基因表达的这些差异,因为Illumina全局筛选阵列显示XDH和PNP中的单核苷酸多态性(SNP)在IBS亚组和HC之间没有差异分布。总之,这些发现提出了一种模型,其中微生物群和宿主的嘌呤核苷酸在结肠组织中诱导代谢应激。 反过来,这可能会通过增加嘌呤挽救而导致代偿性反应。 使用这种多组学观点,我们建议低水平的嘌呤核苷酸可能导致较低的上皮能量状态和粘膜修复能力,这可能部分是IBS的病理生理基础。
研究结论
在这项研究中,我们描述了在不同IBS亚型患者的宿主生理情况下,对肠道微生物组,代谢组,宿主表观基因组和转录组进行综合纵向多组学分析的结果。IBS-D患者活检中基线结肠分泌增加,这表明上皮运输的固有变化或促进液体分泌的代谢产物增加。 观察到的促分泌素(例如主要的BA CDCA和细菌代谢物类胰蛋白酶)的增加表明,较高水平的微生物群相关分泌化合物可能会导致IBS-D分泌增加。 三组结肠活检样本之间对色胺的分泌反应缺乏显着差异,这进一步得到了支持,如果结肠上皮存在固有缺陷,这是可以预期的。
先前的研究表明,BA吸收不良驱动了IBS-D的肠道分泌增加,但是在该研究中,继发性BA与原发性BA并存的增加并没有增加,这表明原发性BA的微生物生物转化减少可能至少部分地驱动了这种作用。通过进一步有针对性地整合多个宿主和微生物组数据层,确定了嘌呤代谢的宿主-微生物途径,这可能在IBS的病理生理中起重要作用。
研究意义
这是首次将次黄嘌呤与IBS发病机制联系起来,包括先前对动物模型进行的致生菌研究。 这说明了在人类中采用多组学测量以鉴定可能依赖于基因表达中人类特异性反应的潜在疾病机制的相关性。由于黄嘌呤氧化酶抑制剂别嘌呤醇(用于治疗痛风)和硫嘌呤(用于优化炎症性肠病治疗)的可用性,次黄嘌呤是有吸引力的药物靶标。
该研究为将来的研究提供了多个新的治疗靶标。IBS-D患者中BA明显减少的微生物生物转化可以使用确定的微生物菌群进行治疗,同样,在IBS-C患者中,细菌SCFA和/或色胺的产生增加可能是可行的治疗策略。
最后,在肠道内局部刺激微生物次黄嘌呤的产生或抑制黄嘌呤氧化酶将是一种增加腔次黄嘌呤的量而没有全身作用的新方法,并且可能对独立于疾病亚型的IBS有益。
研究的局限性
研究存在一些局限性。本文主要关注结肠微生物组,但我们知道小肠可能在IBS症状的产生中起重要作用。 需要专门针对小肠微生物组的纵向研究来补充发现并增进对IBS的理解。
福利活动和技术推广
本文的浅宏基因组测序方案是针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,但是物种的分辨率并没有低于一般宏基因组(普遍5~10G数据量)。
经过几个月的研发的测试,我们推出浅宏基因组测序分析服务,每个样本数据量不低于100万reads,不通过拼接组装,直接基于标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。周期在:2-3周左右,尤其适合粪便样本, 特别推荐。
浅宏基因组分析内容

https://wap.sciencenet.cn/blog-2040048-1251102.html
上一篇:菌群结合临床干预治疗案例分析
下一篇:[转载]Science|慢性肾病与肠道菌群新机制