博文
[转载]JMCB | 张世华课题组提出解决单细胞转录组数据高度缺失及稀疏的新方法
|||

传统的基因测序技术采用“混池”(bulk)测序,得到的是一群细胞中基因表达水平的平均值,掩盖了细胞之间的异质性,这对于理解生物细胞的多样性存在明显的局限性。单细胞转录组测序(single-cell RNA-seq)能够独立地提供每个细胞的基因表达图谱,这样就可以揭示各个细胞间的微小差异,找到单个细胞的独特性,对于我们理解细胞的起源、功能、发育等有着至关重要的作用。
然而,在单细胞层次上,转录组的随机波动会远远大于细胞群体的平均行为,由于每个细胞的mRNA拷贝起始量较低以及测序技术原因,单细胞转录组测序数据通常存在drop-out现象,即很多表达的mRNA没有被捕捉到,导致检测出来的基因表达量为零或者接近零。因此,如何从高度稀疏的单细胞转录组数据中精确地恢复这些缺失的信号,从而促进下游生物信息挖掘,是单细胞转录组数据分析面临的重要挑战。
2020年10月1日,来自中国科学院数学与系统科学研究院的张世华课题组在Journal of Molecular Cell Biology(JMCB)在线发表了题为Imputing single-cell RNA-seq data by considering cell heterogeneity and prior expression of dropouts的工作。该工作基于低秩矩阵填补技术【1,2】和单细胞转录组数据的异质性特征,提出一种新型单细胞转录组数据填补方法PBLR。
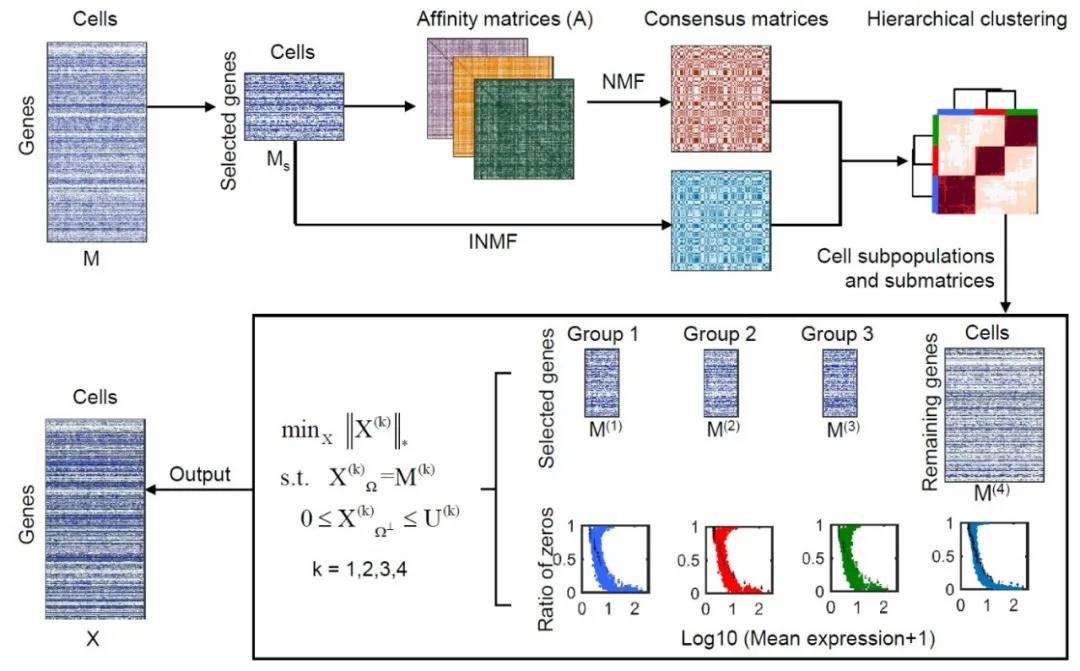
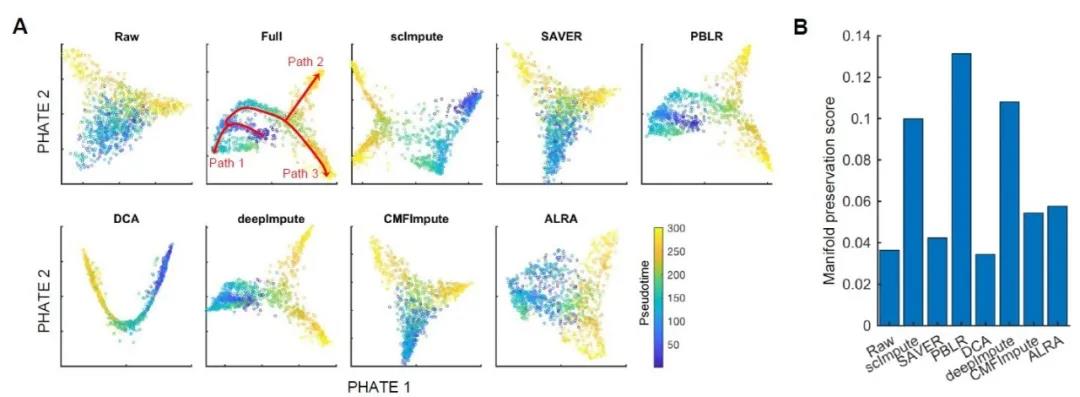
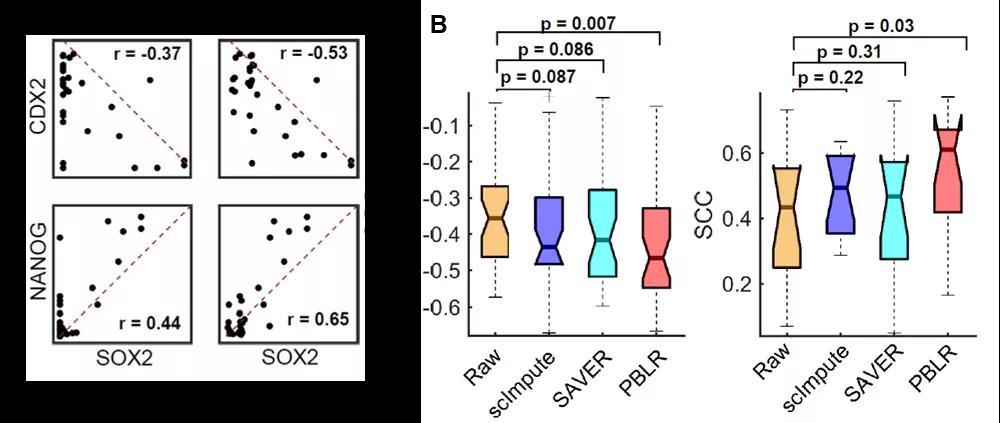
低秩矩阵填补技术在推荐系统中应用广泛,比如著名的Netflix 问题。Netflix公司的电影网站上有非常多的用户,由于大多数用户只评价了部分电影,Nextflix公司希望从已有的评价数据中推测出用户可能喜欢的其他电影。用户的喜爱程度受有限种因素的影响,那么“用户-影片”的评分矩阵是低秩的,故低秩矩阵填补技术被认为是解决该问题的有效方法。类似地,在单细胞转录组数据中,由于细胞种群和共表达基因的存在,数据矩阵也趋向于低秩矩阵,那么低秩填补策略可能适用于单细胞数据的填补问题。 单细胞数据的一大特点是存在很大的异质性,课题组成员敏锐地洞察到这一数据特征对于单细胞数据填补的重要性,决定从细胞子群的角度来解决单细胞转录组数据的drop-out问题。这个想法也可以从电影推荐系统的角度来理解:我们可以把每个基因看成一个电影,每个用户看成一个细胞,不同年龄段的用户对各类电影的喜好程度是不同的。类似地,同一类细胞的基因表达谱有较高的相似度,不同类细胞的基因表达谱有一定的差异。因此,从单细胞数据中识别包含相似细胞的子群,基于细胞子群中基因表达的相似性能够有效地填补缺失值。这一思路也与数学上的基于分块低秩矩阵填补数据具有较高精确性的结论是一致的【3】。另一方面,某个电影的口碑越好,评分越高,看的人越多,那么对于它的评分越多,缺失值就越少。反之,评分低的那些电影,看的人可能相对较少,评分缺失值也就越多。因此,某个电影的评分缺失值在某种程度上与该电影的评分高低有关系。基于此考虑,张世华课题组大胆推测单细胞数据中的drop-out应该也和基因的先验表达有密切关系。此前研究确实也表明表达水平低的基因更倾向于有较大的数据缺失率,而且不同的细胞类型的缺失率不同【4】。因此,利用这个先验关系能够帮助估计出缺失的表达水平的范围。PBLR就是基于细胞子群的有界低秩填补模型,综合考虑了单细胞数据的这几个特点(图1)。 图1. PBLR算法框架 通过PBLR填补后的数据可以很好地用于下游生物信息挖掘。作者采用PBLR分析了8组模拟数据和9组真实数据,结果显示,与主流的6种单细胞数据填补方法相比, PBLR能够更为精确地恢复缺失的数据,能有效提升数据低维可视化、提取基因间关系等下游分析能力(图2和图3)。 图2. PBLR和其他scRNA-seq数据填补算法在模拟数据上恢复缺失数据的比较结果 图3. PBLR恢复真实数据中标记基因之间的相关关系 小结:总结而言,张世华课题组巧妙地类比推荐系统中著名的Netflix问题,充分利用单细胞转录组数据的分布特点,创新性地提出基于细胞子群和基因表达缺失先验信息的单细胞转录组测序数据填补的方法,有效地解决了数据缺失及稀疏的问题、提升了单细胞转录组数据分析的精确性。该方法对于深入挖掘单细胞转录组数据所蕴含的生物医学知识具有重要意义。 参考文献 1. Chen, C.H., He, B.S. and Yuan, X.M. (2012) Matrix completion via an alternating direction method. Ima J Numer Anal, 32, 227-245. 2. Candes, E. and Recht, B. (2012) Exact Matrix Completion via Convex Optimization. Commun Acm, 55, 111-119. 3. Natali Ruchansky, M.C., Evimaria Terzi. (2017) Targeted marix completion. Proc SIAM Int Conf Data Min, doi: 10.1137/1.9781611974973.29. 4. Kharchenko, P.V., Silberstein, L. and Scadden, D.T. (2014) Bayesian approach to single-cell differential expression analysis. Nat Methods, 11, 740-742. 作者简介 张世华,中国科学院数学与系统科学研究院研究员、中国科学院随机复杂结构与数据科学重点实验室副主任、中国科学院大学岗位教授。主要从事生物信息学与机器学习交叉研究,主要成果发表在Advanced Science、National Science Review、Nature Communications、Nucleic Acids Research、Bioinformatics、IEEE TPAIM、IEEE TKDE、IEEE TNNLS、IEEE TFS、AoAS等杂志。目前担任BMC Genomics等杂志编委。曾荣获中国青年科技奖、国家自然科学基金优秀青年基金、国家万人计划青年拔尖人才、全国百篇优秀博士论文奖、中国科学院卢嘉锡青年人才奖等。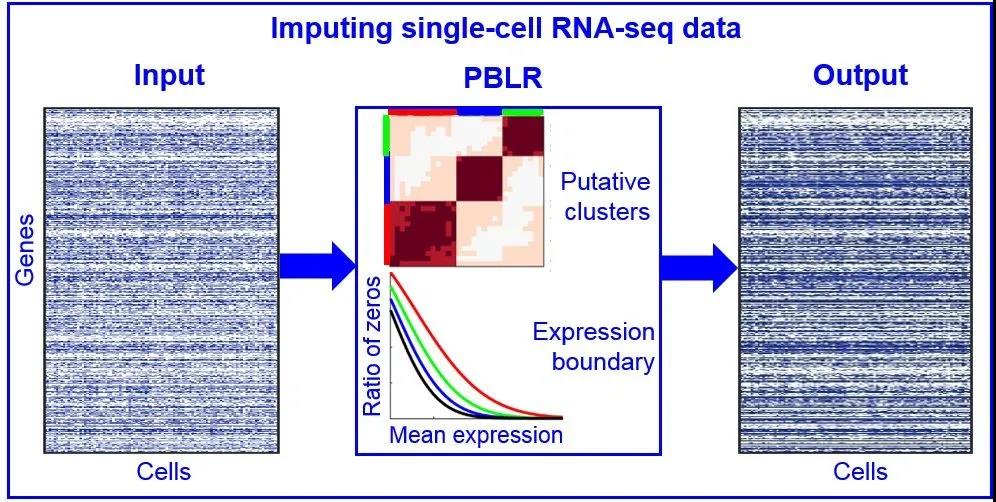


https://wap.sciencenet.cn/blog-1045694-1259022.html
上一篇:[转载]首篇JMCB Application Note│发育生物学工具助力实体瘤可视化研究
下一篇:[转载]JMCB类器官专辑 | 精准搭建人体器官微模型